ADC linker technology plays a critical role in the development of antibody-drug conjugates (ADCs). Acting as the chemical bridge connecting the antibody to a highly potent cytotoxic payload, the design of the linker not only determines ADC stability in circulation but also directly impacts intracellular drug release efficiency, therapeutic efficacy, and safety. As demand for precision-targeted therapies continues to grow, advanced linker technologies are driving the transformation of ADCs from conventional chemotherapy toward highly selective, low-toxicity treatments. This article systematically explores the key types of ADC linkers, design principles, synthetic strategies, and performance optimization methods, while analyzing how stability affects drug-to-antibody ratio (DAR), pharmacokinetics, and off-target toxicity, helping researchers and development teams better understand how optimizing linker technology can enhance ADC efficacy and safety.
ADCs achieve precise delivery of cytotoxic drugs to tumor cells by combining monoclonal antibodies with potent payloads. Linkers play a pivotal role in this process, serving as the core element that governs ADC stability, drug release mechanisms, and overall therapeutic performance. With advances in conjugation chemistry, linker technology has evolved from simple chemical bonding into controllable, intelligent delivery systems, driving ADCs from traditional chemotherapy toward precision-targeted therapies.
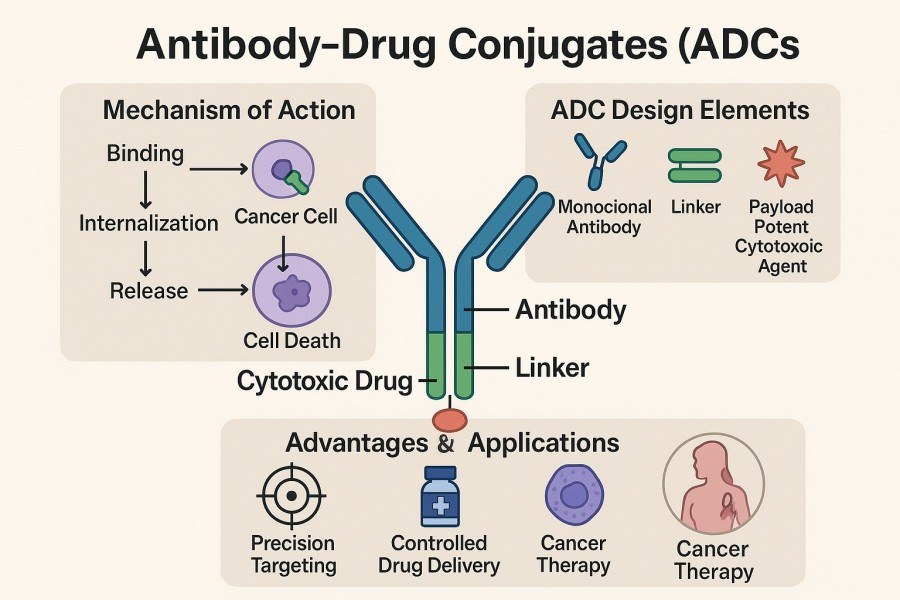 Fig. 1. ADC linker technology (BOC Sciences Authorized).
Fig. 1. ADC linker technology (BOC Sciences Authorized).
An ideal linker should remain chemically inert during circulation to prevent premature drug release and associated toxicity. Once the ADC undergoes receptor-mediated endocytosis into target cells, the linker must respond to intracellular cues—such as acidic pH, elevated glutathione levels, or specific protease activity—to achieve precise drug release. This controlled-release mechanism defines the ADC's therapeutic window, maximizing tumor cell killing while minimizing damage to healthy tissues.
The chemical nature of the linker determines the efficacy, safety, and pharmacokinetic profile of an ADC. Different linker types—such as cleavable and non-cleavable—differ fundamentally in design and mechanism. Cleavable linkers can break under specific physiological conditions to release free drug for cytotoxic action, whereas non-cleavable linkers remain chemically stable, requiring antibody degradation for drug release. These structural differences dictate the intracellular activation pathway of ADCs and their exposure risk to normal tissues. Moreover, chemical parameters such as electronic structure, steric hindrance, and hydrophilicity influence overall pharmacokinetics and plasma half-life. For instance, hydrophobic linkers may induce ADC aggregation or alter antibody conformation, affecting stability, while linkers with hydrophilic groups can enhance solubility and biocompatibility.
Thus, linker chemistry is not only the physical bridge between antibody and payload but also a central design element that balances ADC safety and efficacy.
Diverse linker designs are essential for achieving precise drug delivery. Different linker types determine the triggering mechanism for drug release, stability, and toxicity profile. By selecting appropriate linker chemistries, researchers can achieve an optimal balance between circulatory stability and efficient intracellular payload release. Currently, ADC linkers are primarily categorized into two classes: cleavable and non-cleavable. Additionally, derivative designs based on specific chemical triggers—such as acid-sensitive hydrazones, disulfide bonds, peptide linkers, and self-immolative linkers—offer flexible options for various therapeutic requirements.
Cleavable linkers are among the most common designs, capable of breaking under specific physiological conditions to rapidly release the drug inside target cells. These linkers rely on biological triggers such as acidic environments, enzymatic catalysis, or reductive intracellular conditions. For example, acid-sensitive linkers hydrolyze in lysosomal acidic conditions, while peptide linkers utilize protease-mediated hydrolysis for drug release. In contrast, non-cleavable linkers connect antibodies to payloads via stable covalent bonds (e.g., thioether or alkyne-azide click chemistry), providing excellent plasma stability, with drug release dependent on antibody degradation—minimizing premature off-target effects.
In ADC development, choosing between cleavable and non-cleavable linkers generally depends on the payload's intracellular activation mechanism and target characteristics:
Thus, linker selection is not only a chemical decision but also a strategic design integrating pharmacokinetics and biological mechanisms.
Hydrazone linkers were among the earliest cleavable structures applied in ADCs. Their design exploits the acidic environment inside tumor cells (pH ~5–6), allowing hydrolysis within lysosomes to release the drug. While chemically simple and kinetically controllable, early versions exhibited limited plasma stability, leading to partial degradation in neutral environments and increased toxicity. Recent improvements, such as introducing steric hindrance or electron-donating groups around the hydrazone, have enhanced stability, bringing renewed interest in modified hydrazone linkers in next-generation ADCs.
Disulfide linkers leverage differences in redox environments inside and outside cells to achieve selective drug release. Tumor cells typically contain elevated glutathione (GSH) levels, which reduce disulfide bonds to thiols, triggering drug release. This redox-sensitive mechanism provides excellent targeting specificity, but trace reducing agents in plasma can cause premature cleavage. Modern designs often incorporate steric protection (e.g., methyl or cyclic structures) to enhance selectivity and stability, allowing efficient intracellular activation while maintaining plasma stability.
Peptide linkers are among the most widely used cleavable structures, validated in FDA-approved ADCs such as Adcetris® and Polivy®. They typically contain amino acid sequences recognized by lysosomal proteases (e.g., Cathepsin B, Cathepsin L), such as Val-Cit, Val-Ala, or Gly-Phe-Leu-Gly. Peptide linkers offer high tunability: amino acid sequence, spatial conformation, and amide polarity can be adjusted to control drug release rate and selectivity. Additionally, peptide linkers provide excellent biocompatibility and degradability, making them a standard choice for next-generation ADC development.
Self-immolative linkers represent an advanced stage of linker technology. Once triggered, they automatically decompose, releasing the drug while eliminating residual fragments to ensure the payload acts in a pure form. Typical mechanisms include carbonate, carbamate, or benzyl alcohol self-immolative systems. Multi-stimuli-responsive linkers are emerging, capable of responding simultaneously to pH, redox, electrochemical, or light signals, enabling controlled drug release. These advanced designs significantly improve ADC delivery efficiency and provide greater engineering flexibility for precise release in complex tumor microenvironments.
Explore our wide range of high-quality ADC linker products, including cleavable, non-cleavable, stimuli-responsive, and multifunctional options. Tailored for stability, precise payload release, and research-ready applications.
Designing ADC linkers is not merely a task of chemical synthesis; it is a systematic engineering challenge integrating medicinal chemistry, molecular biology, and pharmacokinetics. An ideal linker must remain stable during circulation while enabling efficient, controllable drug release within target cells. To achieve this, researchers must consider multiple factors in linker design, including stability, selectivity, cleavability, hydrophobicity, length, and conjugation site. Even minor adjustments to these parameters can significantly impact the overall efficacy, safety, and metabolic behavior of an ADC.
Among all ADC linker design principles, stability, selectivity, and cleavability are regarded as the three core criteria:
These three properties are interdependent rather than independent. For example, enhancing stability may reduce cleavability, while increasing cleavability may compromise plasma tolerance. Designers must achieve a dynamic balance through chemical structure optimization and molecular modeling.
A key challenge in ADC development is achieving both high circulatory stability and efficient payload release at the target site. Overly stable linkers may prevent intracellular drug release, reducing efficacy, while overly sensitive linkers may cleave prematurely in the extracellular environment, causing systemic toxicity. Scientists typically achieve this balance through:
This "chemical fine-tuning" strategy enables hierarchical responsiveness: the linker remains stable under neutral conditions but rapidly responds within tumor cells, achieving precision release.
Beyond chemical reactivity, the physicochemical properties of linkers profoundly influence ADC stability, molecular conformation, and pharmacokinetics.
These factors collectively determine the ADC's physical stability, pharmacokinetics, and therapeutic safety margin. Precise control over linker length, hydrophobicity, and conjugation site at the structural level can significantly enhance clinical performance and manufacturing consistency.
High-quality linkers must provide stable, controllable, and scalable chemical structures while being highly compatible with both antibody and drug molecules. To accommodate diverse drug release mechanisms and in vivo stability requirements, researchers have developed multiple synthetic routes and conjugation strategies to enable efficient translation from laboratory to industrial-scale production.
Cleavable and non-cleavable linkers differ significantly in synthetic approach. Cleavable linkers typically employ reactions such as peptide coupling, esterification, condensation, or nucleophilic substitution to construct cleavage sites. These linkers often include enzyme-sensitive sequences (e.g., Val-Cit, Gly-Phe-Leu-Gly) or chemically sensitive groups (e.g., disulfide, hydrazone, carbonate) that break under specific physiological conditions, releasing cytotoxic payloads. In contrast, non-cleavable linkers emphasize structural stability, forming robust covalent bonds via reactions such as maleimide–thiol addition, alkyne–azide click chemistry, or thioether formation. These linkers are largely non-degradable in vivo, with drug release occurring only upon antibody degradation, enhancing plasma stability and reducing off-target toxicity.
In industrial production, the choice of synthetic route depends on the ADC's physicochemical properties, reaction compatibility, and scalability. For example, solid-phase peptide synthesis improves yield and purity for peptide-based linkers, while modular synthetic strategies simplify processes and enhance batch-to-batch consistency.
Click chemistry provides a powerful toolkit for ADC linker development, offering high selectivity, rapid reaction rates, and high yields. Classic copper-catalyzed azide-alkyne cycloaddition (CuAAC) and strain-promoted azide-alkyne cycloaddition (SPAAC) are widely used for connecting antibodies to drug molecules. Compared with traditional conjugation methods, click chemistry offers:
Additionally, other bioorthogonal reactions, such as trans-cyclooctene–tetrazine ligation (TCO–Tetrazine) and oxanorbornadiene–tetrazine reactions, are increasingly applied in ADC linker construction. These reactions enable efficient conjugation without catalysts, providing new chemical platforms for precise and controllable ADC assembly.
Analytical characterization and quality control of linkers are critical in the ADC development process. Due to the structural complexity and multiple functional groups present in linkers, multidimensional analytical techniques are required to ensure purity, structural integrity, reaction efficiency, and stability. Common analytical methods include:
Quality control also encompasses systematic evaluation of DAR, stability testing, and residual reagent analysis to ensure batch consistency and product safety. In commercial ADC production, standardized analytical workflows not only guarantee traceability but also provide critical data for process optimization, regulatory compliance, and long-term stability studies.
Linker stability is one of the core determinants of ADC efficacy, safety, and pharmacokinetics. It governs the timing and location of drug release, affects ADC integrity in circulation, maintains uniform DAR, and ultimately impacts the therapeutic index. If the linker is too fragile, premature payload release in circulation may cause off-target toxicity; if too stable, intracellular drug release may be hindered, leading to reduced activity. Balancing linker stability and responsiveness is therefore a key challenge in ADC design.
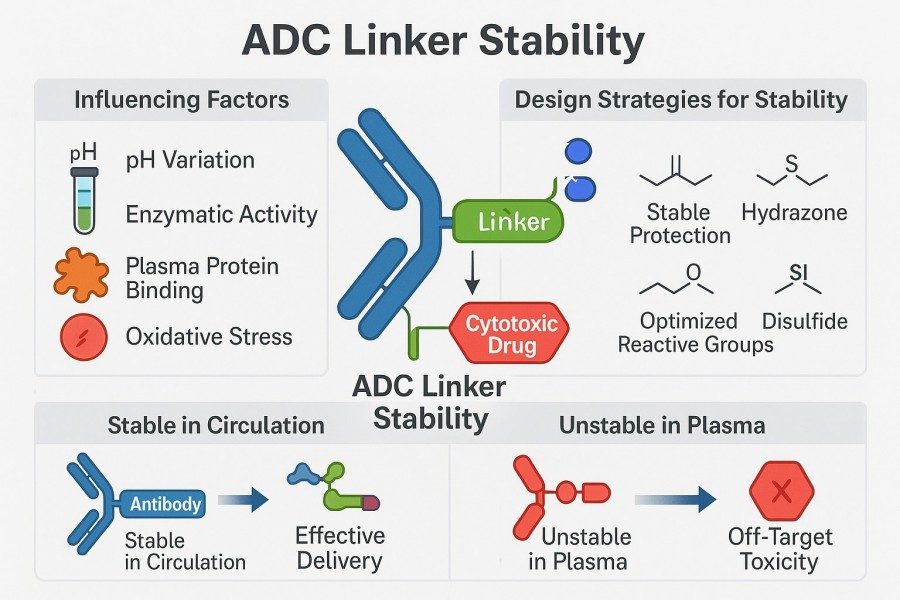 Fig. 2. ADC linker stability (BOC Sciences Authorized).
Fig. 2. ADC linker stability (BOC Sciences Authorized).
Linker stability is closely related to DAR, an important metric for ADC payload loading and consistency that directly affects distribution, metabolism, efficacy, and safety. Unstable linkers may release payloads before reaching target tissue, resulting in decreased DAR, insufficient active dose, and increased exposure to non-target tissues. Conversely, overly stable linkers may trap the drug inside target cells, reducing therapeutic activity. By carefully selecting chemical structures and cleavage mechanisms—such as tuning bond energies, optimizing electronic effects at cleavage sites, and incorporating controllable responsive groups—researchers can maintain DAR stability, achieving optimal pharmacokinetic (PK) profiles and improved drug utilization.
Off-target toxicity is a major safety concern in ADC clinical use. Premature release of cytotoxic payloads can expose healthy tissues, damage hematopoietic systems, and cause unwanted side effects. To mitigate this risk, scientists enhance linker stability to improve in vivo safety. Common strategies include:
These improvements reduce systemic toxicity and extend ADC circulation time, enabling more effective drug accumulation in target cells and higher therapeutic selectivity and safety margins.
In ADC design, extending circulation half-life often conflicts with efficient intracellular drug release. Excessive stability prolongs circulation but may hinder release at target cells, while overly rapid release shortens half-life and reduces tumor-targeting effects. Achieving an optimal therapeutic balance requires linkers with tunable responsiveness. Modern ADC linker designs achieve dynamic control of half-life and release rate through:
By embedding these intelligent responsive units in linker structures, researchers can extend ADC circulation time while ensuring efficient drug release at tumor sites, maximizing efficacy and minimizing side effects.
Despite significant progress over the past decade, ADC linker development still faces multiple challenges. These issues span chemical and biological principles and directly affect clinical efficacy and safety. Successful high-performance linker development requires optimization and balance in stability, targeted release, antibody compatibility, and analytical characterization.
A core challenge is maintaining sufficient stability in circulation while enabling efficient intracellular drug release. Plasma contains enzymes, oxidants, and small reactive molecules that may prematurely cleave the linker, causing off-target toxicity. Conversely, overly stable linkers reduce drug release efficiency in target cells, compromising anti-tumor efficacy. Strategies to address this challenge include:
Another key challenge is ensuring linker compatibility with various antibody structures and cytotoxic payloads. Differences in antibody spatial conformation, glycosylation patterns, cysteine/lysine distribution, and conjugation sites, as well as payload polarity, hydrophobicity, chemical stability, and active form, can all impact conjugation. Incompatibility may lead to:
Linker design must therefore integrate antibody engineering, payload physicochemical properties, and conjugation strategy. Site-specific conjugation, PEGylation, or modular linker designs can support multi-antibody, multi-payload platforms. This approach enhances ADC consistency and supports personalized therapy development.
High-quality ADC production requires precise analysis and characterization, but the chemical complexity of linkers presents analytical challenges:
To address these challenges, researchers often use multidimensional analytical approaches, including LC-MS/MS, HPLC, NMR, SEC, UV-Vis, and biophysical characterization techniques (e.g., DSC, DLS). Standardized data management and batch tracking are crucial to ensure reproducibility in industrial-scale production.
As ADC clinical development and commercialization advance, optimizing linker technology has become a critical factor for improving ADC efficacy, safety, and manufacturability. Best practices encompass not only chemical design and structural optimization but also drug–antibody compatibility, controlled release strategies, and advanced analytical characterization. By systematically integrating these factors, ADCs can achieve higher therapeutic indices and improved batch-to-batch consistency.
Structure–activity relationship (SAR) analysis is a core principle in linker optimization. By evaluating the relationship between linker molecular structure and drug release efficiency, plasma stability, and toxicity, researchers can achieve precise design:
SAR analysis supports data-driven linker design through experimental results and computational modeling, maximizing efficacy while minimizing off-target toxicity.
Optimizing linkers requires balancing plasma stability with intracellular drug release kinetics:
This strategy ensures ADC integrity in circulation while allowing rapid activation within target cells, achieving high therapeutic selectivity and maximal efficacy.
ADC performance heavily depends on payload–antibody compatibility and conjugation site engineering:
A well-designed conjugation strategy not only ensures ADC stability but also improves plasma half-life and target accumulation efficiency.
Modern ADC optimization emphasizes biodegradable and stimuli-responsive linker design:
These innovations enhance ADC selectivity and therapeutic index, providing reliable solutions for precise delivery in complex tumor microenvironments.
Optimizing linker technology relies on precise analytical characterization. Advanced methods allow comprehensive evaluation of structural integrity, drug loading, and functional performance:
Multidimensional analysis enables closed-loop optimization from design, synthesis, performance evaluation, to quality control, ensuring each ADC batch achieves consistent clinical performance and high safety.
BOC Sciences offers end-to-end technical capabilities from early R&D to clinical production, providing efficient and customized ADC linker solutions. Our team combines advanced chemical synthesis, structural modification, performance optimization, and analytical characterization to enhance ADC stability and controlled release in vitro and in vivo, delivering reliable technical support and innovative solutions for drug development.
From cytotoxins to linkers, explore our cutting-edge products for your ADC project.
| Catalog | Name | CAS | Price |
| BADC-01016 | PC Biotin-PEG3-alkyne | 1869922-24-6 | Bulk Inquiry |
| BADC-01018 | Mc-Val-Ala-PAB | 1870916-87-2 | Bulk Inquiry |
| BADC-01021 | Fmoc-NH-PEG2-NH2 | 190249-87-7 | Bulk Inquiry |
| BADC-01182 | THP-SS-alcohol | 877864-04-5 | Bulk Inquiry |
| BADC-01232 | Biotin-PEG3-aldehyde | 889443-90-7 | Bulk Inquiry |
| BADC-01525 | Folate-PEG3-amine | 710323-40-3 | Bulk Inquiry |
| BADC-01646 | Mal-PEG2-Val-Cit-PABA | 1662687-83-3 | Bulk Inquiry |
| BADC-01679 | Mal-PEG2-Val-Cit-PABA-PNP | 1345681-52-8 | Bulk Inquiry |
| BADC-01745 | MC-Val-Cit-PAB-NH-C2-NH-Boc | 1616727-22-0 | Bulk Inquiry |
| BADC-00953 | Ald-CH2-PEG5-azide | 1446282-38-7 | Bulk Inquiry |
Explore our advanced tools and expertise for next-generation ADC research and development.
Potent cytotoxins designed for targeted antibody-drug conjugate therapy.
Stable, selective linkers enabling precise drug release in ADCs.
Integrated payload-linker solutions for efficient and targeted ADC delivery.
Targeted therapeutics combining antibodies with cytotoxic drugs for precision treatment.











 Loading......
Loading......