ADC linker is the bridge between antibodies and cytotoxic drugs and plays a key role in antibody-drug conjugate (ADC) drugs because its properties greatly affect the therapeutic indicators, efficacy, and pharmacokinetics of these drugs. The ideal conjugation must be stable in vitro or in the blood circulation to prevent systemic toxicity caused by premature release of cytotoxic drugs, while at the same time being able to enter and kill cancer cells through the rapid release of effective cytotoxic drugs. BOC Sciences can provide ADC linkers with multiple cleavage mechanisms according to your project needs, including enzymatic cleavage linkers, chemical cleavage linkers and peptide linkers. We also support the integrated design of linkers to modulate payload release and ADC stability for optimal efficacy of ADC drugs.
Comprehensive one-stop antibody-drug conjugate service platform
Large Stock
More than 1000+ high-purity products in inventory

Global Delivery
Warehouses in multiple cities to ensure fast delivery

mg to kg
Qualified facilities & equipment of cGMP laboratory

24/7 Technical Support
Strict process parameter control to ensure product quality

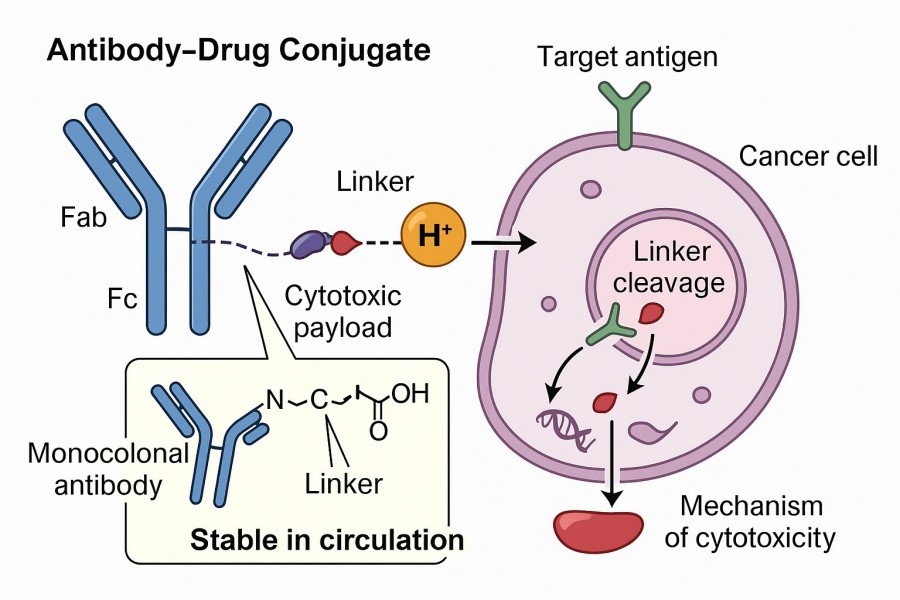
The basic structure of an ADC includes a monoclonal antibody, a cytotoxic small-molecule drug (payload), and a linker. Among these components, the linker is an essential element in ADC design, connecting the antibody to the cytotoxic payload through covalent coupling. The ADC linker should be stable enough in the circulation to maintain the ADC drug concentration in the blood circulation and not be released before the cytotoxic drug reaches the target, thereby causing minimal off-target effects and improving the safety of the ADC drug. At the same time, appropriate hydrophilicity or lipophilicity of the linker can enhance payload coupling and reduce immunogenicity properties and is also a key aspect of the linker. Therefore, in the design of ADC drugs, these important parameters of the linker must be correctly adjusted to achieve a balance between ADC stability and payload release efficiency, in order to achieve the expected effects of ADC drugs.
In ADC design, the type and properties of the linker are key factors that determine whether the drug can achieve clinical success. The linker not only controls how the payload is released but also affects the ADC's blood stability, half-life, selectivity, and overall safety. Currently, ADC linkers can be broadly classified into two categories: cleavable linkers and non-cleavable linkers. These linkers play an important role in determining the pharmacokinetic properties, selectivity, therapeutic index, and overall success of ADCs. Both types of linkers have been shown to be safe in preclinical and clinical trials.
Cleavable linkers are the most widely used type and primarily rely on specific conditions within tumor cells or the tumor microenvironment to trigger payload release. The linker may be cleavable. Here, a chemical bond (or multiple chemical bonds) between the payload and the antibody attachment site (usually an amino acid) is cleaved intracellularly. The cleavable linker can be degraded under different pH conditions or by the action of intracellular enzymes to achieve separation of the drug from the antibody. However, since the drug may diffuse away from the target cell after release, it can also affect adjacent tissues. Cleavable linkers generally include enzyme-sensitive linkers, pH-sensitive linkers, and redox-sensitive linkers. The advantage of this type of linker is that it enables efficient, rapid, and controllable payload release in the target environment, but its design must also ensure stability in circulation; otherwise, premature toxicity may occur.
Non-cleavable linkers fall into two groups, thioether or maleimidocaproyl (MC). They consist of stable bonds that protect against proteolytic cleavage and ensure greater plasma stability than their cleavable counterparts. ADCs containing such linkers rely on complete lysosomal enzymatic degradation of the antibody, releasing the payload upon internalization, resulting in simultaneous detachment of the linker. The advantage of non-cleavable linkers over cleavable linkers is their improved stability. Genentech/Immunogen has successfully explored this linkage strategy, with Trastuzumab emtansine (Kadcyla/T-DM1) gaining clinical approval. This ADC contains a non-cleavable SMCC (N-succinimidyl-4-(maleimidomethyl)cyclohexane-1-carboxylate) linker to link warhead DM1 cytotoxin to an anti-HER2 mAb Lys residue of Trastuzumab. The drug conjugate exhibits greater activity than traditional trastuzumab-DM1 or trastuzumab conjugated to other maytansinoid alkaloids via reducible disulfide bonds.
Linkers can be classified based on their drug release mechanisms and their stability in circulation. In the past few years, many new linkers have been developed, including cathepsin-cleavable linkers, acid-cleavable linkers, GSH-cleavable linkers, Fe(II)-cleavable linkers, novel enzyme-cleavable linkers, photoresponsive cleavable linkers and bioorthogonal cleavable linkers. Among them, cathepsins, GSH, and acid-cleavable linkers have been well studied and used in approved ADCs.
Enzyme-sensitive linkers rely on lysosomal proteases that are specifically overexpressed within tumor cells for cleavage. A common example is the Val-Cit (valine-citrulline) linker, which is cleaved by Cathepsin B in lysosomes, subsequently releasing the potent small-molecule toxin MMAE (Monomethyl Auristatin E). This design takes full advantage of the enzymatic environment inside tumor cells, enabling efficient drug release once the ADC enters the cell, while maintaining stability in the bloodstream. A typical representative is Brentuximab Vedotin (Adcetris®), which achieves precise targeted delivery through the Val-Cit-PAB linker, serving as a successful example in the ADC field. The advantage of enzyme-sensitive release mechanisms lies in their high selectivity and release efficiency, but differences in enzyme expression across tumor tissues may affect therapeutic efficacy.
| Linker Types | Description |
| Cathepsin B Cleavable Linker | The first category is peptide linkers, which mainly include dipeptide linkers and tetrapeptide linkers. The mechanism of peptide linker cleavage is that after internalization of ADC through endocytosis and transport to lysosomes, cathepsin B selectively cleaves the linker, thereby releasing the payload. The most commonly used dipeptide linkers among marketed ADC drugs include Val-Cit and Val-Ala dipeptides. The stability and cell activity of the two linkers are equivalent. In addition to dipeptide linkers, the tetrapeptide Gly-Gly-Phe-Gly has also been successfully used in ADC drugs. Compared with dipeptides, tetrapeptide linkers are more stable in blood circulation, and the marketed ADC drug Enhertu uses this type of linker. |
| β-Glucuronide Linker | β-glucuronidase-sensitive ADCs are covalently bound to cytotoxic drugs and antibodies by combining a β-glucuronidase linker with a self-eliminating PABC spacer molecule. It releases the payload through enzymatic cleavage by β-glucuronidase, an enzyme commonly found in the tumor microenvironment, which enhances targeted drug delivery in ADCs. |
| β-Galactosidase Cleavable Linker | Similar to β-glucuronidase, β-galactosidase is overexpressed in certain tumors, where it hydrolyzes β-galactosidic bonds, releasing the drug. The difference is that β-galactosidase only exists in lysosomes, while β-glucuronidase is expressed in lysosomes and also in the microenvironment of solid tumors. |
| Sulfatase-Cleavable Linker | A third type of enzymatically cleavable linker is sulfatase cleavable linkers. Sulfatases are overexpressed in multiple cancer types, exhibiting potential selectivity. The study involved Her2 antibodies with MMAE as the payload. Compared with the classic cleavable Val-Ala and Val-Cit linkers, the sulfatase linker showed similar potency on HER2+ cell lines. |
| Phosphatase-Cleaved Linker | The final category is phosphatase-cleaved linkers. Phosphatases belong to another important class of enzymes that cleave connexins and are target enzymes expressed exclusively in the lysosomal compartment. These linkers target pyrophosphatase and acid phosphatase, which hydrolyze the pyrophosphatase and terminal monophosphate into their respective alcohols, thereby releasing the payload. |
Another common payload release mechanism is based on the acidic environment of tumor cells and lysosomes. Normal blood pH is typically around 7.4, whereas the intracellular environment of tumor cells and lysosomes is generally lower (around 5–6). This difference provides the trigger condition for pH-sensitive linkers. The most typical chemical structure is the hydrazone bond. Hydrazone linkers (a type of acid-labile linker) are highly sensitive to acidic conditions in the body and use the acidic environment of endosomes and lysosomes to trigger cleavage and subsequent payload release. For example, the early ADC drug Gemtuzumab ozogamicin (Mylotarg®) utilized a hydrazone linker to achieve selective payload release under acidic conditions. The advantage of pH-sensitive mechanisms lies in their simple design and mature synthetic pathway, allowing effective exploitation of the natural differences in the tumor microenvironment.
In addition to enzymatic and pH conditions, another type is the reducible disulfide bond linker. Disulfide bonds depend on reduced glutathione. Compared with plasma (∼5 μmol/L), glutathione levels in the cytoplasm are higher (1–10 mmol/L). Therefore, the reduced disulfide linker remains relatively stable in blood circulation, while intracellular glutathione reductively cleaves the disulfide linker to release the payload. To further enhance selectivity, researchers often introduce substituents such as methyl groups around the disulfide bond to adjust the cleavage rate. The advantage of redox-sensitive linkers lies in their rapid release and high selectivity, making full use of the unique metabolic environment of tumor cells.
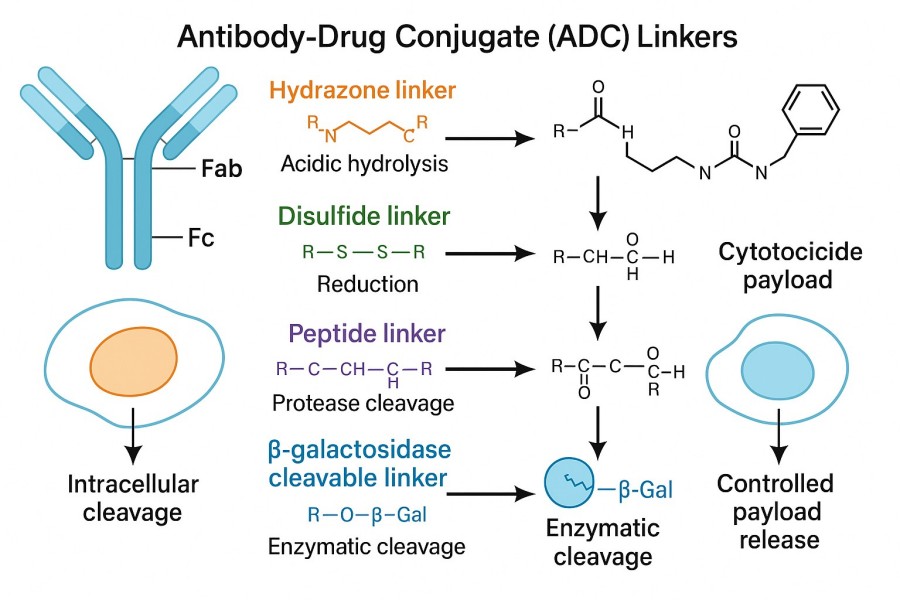
ADC linker design not only relies on biological requirements but also heavily depends on chemical strategies. The chemical properties of the linker directly determine the binding strength, stability, and in vivo release profile of the drug molecule. Therefore, a deep understanding of linker chemistry is a core aspect of ADC development and commercialization. The linker is composed of three parts: isolation rod, cleavage zone and release zone. Among them, the cleavage zone is the core part of the linker, and the cleavage site of the linker is usually in the cleavage zone. The function of the isolation rod and the release zone is mainly to modify the hydrophilic/hydrophobicity of the linker. Commonly used modifiers include MC (maleimidocaproyl), PEG (polyethylene glycol) and PABC (p-aminobenzyl alcohol). The release zone will be released together with the toxin after the linker is broken, so the release zone can also modify the hydrophilicity/hydrophobicity of the toxin after the linker is broken. The ideal ADC linker is mainly reflected in:
The core chemistry of ADC linkers lies in their functional groups, which enable efficient conjugation between the antibody and the drug molecule. Common bonding strategies include forming thioether bonds through thiol (–SH) groups reacting with maleimide groups, or forming amide bonds via amino (–NH2) groups reacting with carboxyl or activated ester groups. These chemical reactions require not only high selectivity but also mild reaction conditions to avoid damaging the antibody structure or affecting drug activity. As research progresses, more controllable cleavable bonds, such as disulfide and oxime bonds, have been introduced, allowing the linker to break under specific conditions for more precise drug release. Additionally, incorporating PEG segments into the linker can significantly enhance the solubility and biocompatibility of ADC molecules while reducing aggregation caused by hydrophobicity. PEG chains also act as "spacers," increasing the spatial flexibility between the antibody and the drug, thereby improving accessibility to the drug's binding sites.
Enhances ADC solubility and stability by introducing polyethylene glycol for improved pharmacokinetics.
Provides selective conjugation with thiol groups, forming stable bonds under physiological conditions.
A maleimidocaproyl linker offering high stability and controlled payload release in ADCs.
Composed of enzyme-sensitive peptides, enabling tumor-specific payload release in ADCs.
A disulfide-based cleavable linker facilitating payload release in reductive tumor environments.
A valine-citrulline dipeptide linker cleaved by cathepsins in tumor cells for selective drug release.
A heterobifunctional linker enabling thiol-to-amine crosslinking with excellent stability and reactivity.
Utilizes amino acids for precise payload attachment and biodegradable linkage in ADCs.
The stability of ADC linkers is a fundamental aspect determining the therapeutic efficacy and safety of ADCs. Linkers are designed to attach cytotoxic drugs to antibodies and play a key role in controlling when and where the payload is released. A stable linker ensures that the ADC remains intact during systemic circulation, preventing premature drug release that could lead to off-target toxicity. Cleavable linkers are designed to exploit specific conditions in the tumor microenvironment, such as acidic pH, elevated glutathione levels, or specific enzymes like cathepsins, to trigger drug release. Non-cleavable linkers, on the other hand, remain stable until the ADC is internalized by target cells, where lysosomal degradation releases the drug. Achieving an optimal balance between stability and timely payload release is essential for ADC development. A well-designed linker ensures precise delivery of the drug to tumor cells while minimizing systemic side effects, thereby enhancing the therapeutic index of ADCs.
In ADC development, linker design is a core element for achieving efficient and precise drug delivery. An ideal ADC linker must ensure payload stability in vivo while enabling precise release under specific conditions in the tumor microenvironment or inside cells, thereby maximizing therapeutic efficacy and minimizing systemic toxicity. Scientifically rational linker design principles directly determine an ADC's pharmacokinetics, tumor selectivity, and clinical performance.
Linker design must balance blood stability with payload release efficiency. If the linker is too stable, the payload may be insufficiently released within tumor cells, reducing anti-tumor activity. Conversely, if the linker is unstable, premature payload release in circulation can increase off-target toxicity. Therefore, design must consider the type of chemical bonds, reactivity of functional groups, and microenvironment-triggered mechanisms, ensuring the ADC remains intact until it reaches the target and efficiently releases the payload at the tumor site.
The chemical characteristics of linkers significantly affect ADC pharmacokinetics (PK) and biodistribution. Stable and hydrophilic linkers typically prolong circulation time and enhance tumor accumulation, whereas highly hydrophobic or easily cleavable linkers may cause early degradation or nonspecific distribution, increasing the risk of side effects. Additionally, linker length and structure influence the spatial conformation between antibody and drug, altering tissue penetration and targeting efficiency. Therefore, linker design should integrate PK/PD (pharmacokinetic/pharmacodynamic) data to optimize in vivo distribution and drug exposure.
The synthesis and manufacturing of ADC linkers are key steps for producing high-quality drugs. The chemical complexity of linkers and their biocompatibility requirements mean that their synthesis involves not only precise chemical reaction design but also consideration of scalability and process control. Throughout ADC development, linker synthesis strategies, large-scale production, and custom manufacturing capabilities are critical factors affecting product consistency and efficacy.
ADC linker synthesis typically relies on precisely controlled organic reactions, such as amidation, thioether formation, esterification, and click chemistry. Amidation and thioether formation are used to construct stable antibody-drug bonds, while click chemistry and bioorthogonal reactions enable site-specific conjugation to ensure consistent drug-to-antibody ratio (DAR). For cleavable linkers, the incorporation of dipeptides, hydrazone bonds, or disulfide bonds requires multistep synthesis under strictly controlled conditions to ensure stability in plasma and efficient release in target environments. Each reaction step's yield, purity, and reproducibility directly impact final ADC performance; therefore, linker synthesis often involves quality control using HPLC, mass spectrometry (MS), and nuclear magnetic resonance (NMR).
Scaling laboratory-synthesized linkers to industrial production presents multiple challenges. Certain reaction conditions may be controllable on a small scale but can result in side reactions or reduced yield at larger scales. Linkers are often sensitive to temperature, pH, and solvent conditions, requiring strict process control. Industrial production must also comply with GMP standards to ensure product purity and biocompatibility while meeting solvent compatibility and solubility requirements for antibody conjugation. These factors make industrial-scale linker production a critical challenge in ADC development.
The latest ADC linker technologies focus on intelligent, multi-responsive, and controllable payload release, with site-specific conjugation ensuring precise DAR to improve ADC efficacy and consistency. PEG spacers enhance solubility, stability, and tissue distribution while improving antibody-drug binding efficiency. Advanced analytical tools such as HPLC, MS, NMR, and DAR measurement allow precise assessment of linker purity, structural integrity, and conjugation efficiency, providing full-process quality control from laboratory synthesis to industrial production and reliable data support for ADC development and clinical translation.
BOC Sciences combines advanced chemistry platforms with deep ADC expertise to support your linker design and optimization. From cleavable and non-cleavable linkers to custom synthesis and tailored conjugation strategies, we provide comprehensive solutions to enhance your ADC efficacy and safety.
The core of ADCs lies in effectively conjugating the antibody and drug through the linker to achieve precise delivery. Linker-payload conjugation efficiency and the DAR directly determine ADC activity, selectivity, and safety. Optimizing conjugation strategies and controlling DAR are critical steps for achieving high-performance ADCs.
In ADC design, the chemical compatibility between the linker and the payload is critical. Different payloads have varying hydrophobicity, chemical reactivity, and stability, making the choice of appropriate functional groups and chemical bonds essential for successful and stable conjugation. For example, highly hydrophobic toxins such as MMAE or maytansinoids often require PEGylation or hydrophilic spacer arms to reduce aggregation risk. At the same time, the linker structure must remain stable in circulation while efficiently releasing the payload under tumor microenvironment or intracellular conditions, ensuring both efficacy and safety. Proper design enables the formation of an efficient and stable conjugation system, providing superior pharmacokinetic and pharmacodynamic properties for ADCs.
The DAR is a key quality metric for ADCs, reflecting the number of payload molecules conjugated per antibody. A high DAR can cause antibody aggregation, reduce circulation stability, and increase nonspecific toxicity, whereas a low DAR may compromise anti-tumor activity. Modern ADC development often achieves precise DAR control through site-specific conjugation or optimized conjugation chemistry. For instance, highly selective reactions such as thiol–maleimide or click chemistry can produce uniform DAR distributions, significantly improving ADC efficacy, consistency, and safety.
Linkers not only affect payload release but also directly determine ADC safety, drug delivery efficiency, and selectivity. A well-designed linker ensures efficient payload release within target cells while minimizing off-target release in circulation. Conversely, unstable or incompatible linkers can cause systemic toxicity and reduce the therapeutic window. Linker structure also influences antibody conformation and pharmacokinetics, altering ADC distribution and half-life. Therefore, designing a scientifically optimized linker-payload conjugation system and controlling DAR are key steps for achieving safe and effective therapy, providing a significant advantage in both clinical and commercial ADC development.
Selecting the right linker is a critical step in ADC development, determining drug delivery efficiency, targeting, and safety. A high-quality linker must match the chemical characteristics of the payload while considering the antibody structure, in vivo stability, DAR control, and clinical and manufacturing requirements. A scientific evaluation of selection criteria and matching strategies is essential for accelerating ADC development, enhancing efficacy, and minimizing risk.
When selecting a linker, key evaluation criteria include stability, cleavability, chemical compatibility, and biocompatibility. A stable linker ensures the ADC remains intact in circulation, reducing off-target toxicity. A cleavable linker must efficiently release the payload under specific tumor microenvironment or intracellular conditions. Chemical compatibility ensures efficient conjugation with both payload and antibody while maintaining protein integrity. Biocompatibility involves PEGylation, hydrophilic modifications, and immunogenicity control, providing safety for clinical applications.
Linker selection must also consider the chemical and structural properties of the payload and antibody. Hydrophobic payloads often require hydrophilic spacer arms or PEGylation to improve solubility and circulation stability. Enzyme-sensitive or pH-sensitive linkers are suitable for releasing payloads under specific tumor conditions. The antibody's thiol or amino group positions determine the conjugation method, such as thiol–maleimide or amino–activated ester coupling. Proper matching of linker, payload, and antibody structure is key to achieving efficient conjugation, uniform DAR, and superior efficacy.
Beyond scientific matching, linker selection must also account for industrial and regulatory factors. Industrial-scale production requires linkers that are scalable, high-purity, and easy to control, with good stability and shelf life. Commercial considerations include cost, raw material supply, and customization capability. Regulatory requirements involve GMP compliance, drug consistency, safety evaluation, and clinically supported conjugation methods. By integrating scientific, industrial, and regulatory factors, development teams can select the optimal linker solution to ensure smooth translation from laboratory research to clinical application.
ADC linkers function primarily by controlling payload release in vivo to achieve precise drug delivery. Cleavable linkers break under specific conditions, such as enzyme-sensitive linkers cleaved by lysosomal enzymes, pH-sensitive linkers cleaved in acidic environments, and redox-sensitive linkers cleaved by high intracellular glutathione in tumor cells. Non-cleavable linkers release payload slowly through antibody degradation within target cells. Different linker mechanisms balance circulation stability with targeted release, enhancing efficacy while reducing off-target toxicity.
A payload linker is the chemical bridge that connects a small-molecule toxin to the antibody. It ensures payload stability in circulation while enabling release under specific conditions in target cells for precise therapy. Payload linkers are typically cleavable or non-cleavable, responding to enzymes, pH, or reductive conditions, or designed for extended circulation. Thoughtful design of payload linkers is critical for ADC safety, efficacy, and consistency.
In ADCs, the linker serves as both a bridge and regulator. It chemically connects the antibody and payload, enabling precise toxin delivery to tumor cells while maintaining stability in circulation. The linker also determines the payload release rate and location, affecting pharmacokinetics, efficacy, and off-target toxicity. Choosing the right linker type and structure optimizes payload release, DAR, and tumor selectivity, making it a crucial component of ADC design and clinical application.
ADC linkers are mainly classified into cleavable, non-cleavable, and hybrid types. Cleavable linkers break under enzymes, acidic environments, or reductive conditions to release the payload in target cells. Non-cleavable linkers release toxins slowly through antibody degradation, offering higher stability. Hybrid or next-generation linkers combine multi-responsive mechanisms and site-specific conjugation to optimize payload release and DAR consistency. Each type offers distinct advantages in stability, release efficiency, and targeting.
An ADC linker payload is the cytotoxic drug attached to an antibody via a chemical linker. The linker ensures stability in circulation and targeted release inside cells. Proper linker-payload design maximizes therapeutic efficacy, reduces off-target toxicity, and maintains a consistent DAR for reliable ADC performance.
An ADC linker stability test assesses how well a linker maintains integrity under physiological conditions. Using methods like HPLC, LC-MS, and incubation studies, researchers monitor payload release and linker durability, ensuring the ADC remains effective in circulation while minimizing premature release and systemic toxicity.
ADC linker design balances circulation stability with efficient payload release. It considers cleavable vs non-cleavable linkers, functional groups, PEGylation, enzyme or pH sensitivity, and site-specific conjugation to control DAR, improving ADC efficacy, selectivity, and safety.
From cytotoxin synthesis to linker design, discover our specialized services that complement your ADC projects.
Find exactly what your project needs from our expanded range of ADCs, offering flexible options to fit your timelines and goals.
![]()
![]()
![]()
![]()
![]()











 Loading......
Loading......