ADC linkers are the key chemical structures in antibody–drug conjugates (ADCs) that connect monoclonal antibodies with cytotoxic drugs (payloads). They not only affect the stability of the drug in vivo but also determine the manner and timing of active payload release. According to different release mechanisms, ADC linkers can be divided into two major categories: cleavable and non-cleavable. Cleavable linkers can break under specific biochemical conditions inside target cells, enabling rapid drug release; non-cleavable linkers rely on antibody degradation to release the drug and maintain higher stability in circulation. Understanding the structural characteristics, release mechanisms, and clinical application differences of these two types of linkers is of great significance for optimizing ADC design, improving efficacy, and ensuring safety.
ADCs combine the high specificity of monoclonal antibodies in recognizing tumor cells with the potent killing power of small-molecule cytotoxic drugs, introducing the concept of "targeted delivery" into chemotherapy drug delivery. This greatly enhances therapeutic efficacy while reducing damage to normal tissues. Unlike traditional chemotherapy, ADCs can remain stable in the bloodstream until they precisely reach target cells, where they then release their lethal payload. This design not only improves the therapeutic index but also significantly reduces systemic toxic side effects. Among the three core components of ADCs—antibody, drug, and linker—the linker is the key element ensuring "precise release." Located between the antibody and the drug, it performs a dual function of stable transportation and controlled release. Based on the drug release mechanism, linkers can be divided into cleavable and non-cleavable types.
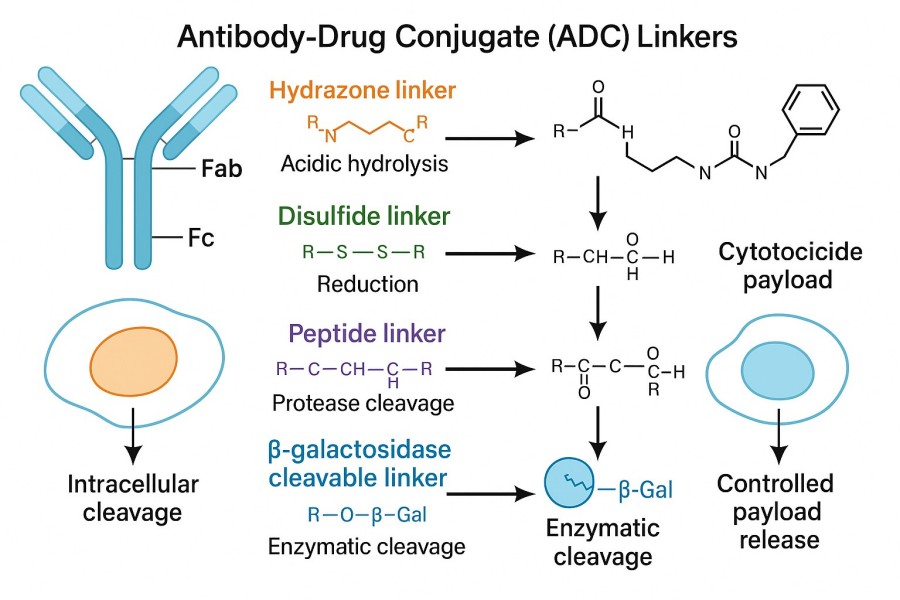 Fig. 1. ADC linker types (BOC Sciences Authorized).
Fig. 1. ADC linker types (BOC Sciences Authorized).
A linker is not merely a physical "bridge" connecting the antibody and the drug—it is a molecular switch that determines when, where, and at what rate the drug will be released. An ideal linker should:
Different types of linkers can significantly affect the pharmacokinetics (PK), biodistribution, therapeutic effects, and safety of ADCs. Cleavable linkers can break rapidly in the special environment inside target cells, releasing the drug to achieve efficient killing—making them especially valuable in strategies requiring fast drug release. Non-cleavable linkers, on the other hand, maintain high stability throughout systemic circulation, with drug release depending on the natural degradation of the antibody. This design generally helps reduce free drug levels in the blood, thereby lowering off-target toxicity. Studies have shown that striking a balance between efficacy and safety requires choosing the appropriate linker type based on payload characteristics, tumor biology, and patient-specific factors. Therefore, linker selection is not only a consideration in chemical synthesis but also an important integration of pharmacology and clinical strategy.
Different linker types exhibit distinct variations in chemical structure, stability, and functional design. These differences not only determine the stability of the ADC in circulation but also influence its release efficiency within the tumor microenvironment.
The core feature of cleavable linkers is the presence of chemical structures that respond to specific biological conditions, such as peptide bonds (e.g., Val-Cit sequence) recognized by lysosomal proteases like Cathepsin B, hydrazone bonds sensitive to acidic environments, and disulfide bonds that can be reduced under reductive conditions. These functional groups remain relatively stable in circulation but are rapidly cleaved under specific intracellular conditions to release the drug. Their chemical design must balance stability and sensitivity to ensure the drug is not released at non-target sites.
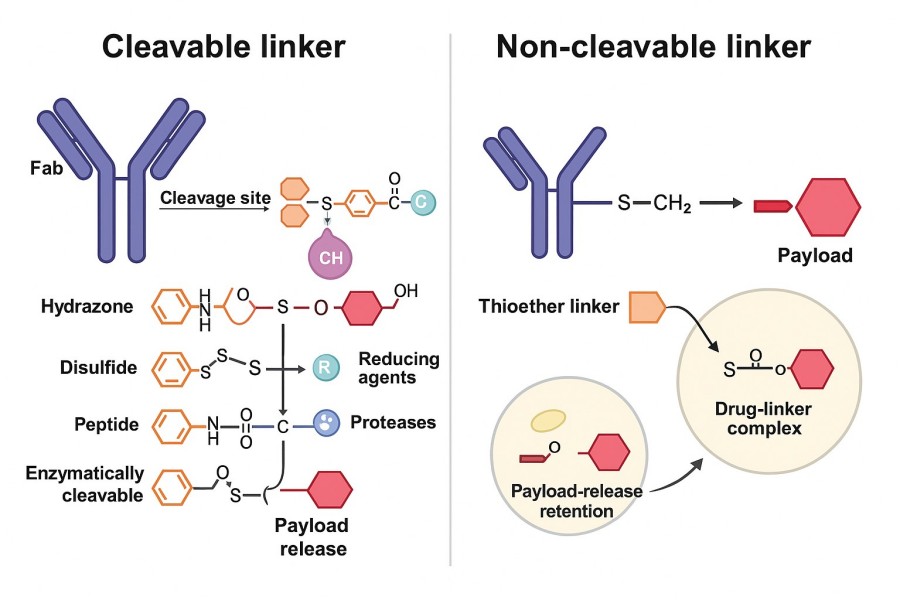 Fig. 2. Difference between cleavable and non-cleavable linkers (BOC Sciences Authorized).
Fig. 2. Difference between cleavable and non-cleavable linkers (BOC Sciences Authorized).
Non-cleavable linkers typically employ highly stable covalent bonds such as thioethers and triazoles, which do not break even under acidic, reductive, or enzymatic conditions. These linkers rely on intracellular antibody degradation to release the drug, focusing on forming a strong and stable bond between the antibody and the drug to minimize non-specific release risk.
In drug development, the design goal for cleavable linkers is to achieve dual characteristics: long-term stability in plasma to prevent premature leakage, and rapid cleavage under specific conditions within tumor cells to ensure precise payload release. This is often achieved by adjusting chemical configurations or shielding reactive sites to enhance selectivity.
Non-cleavable linkers maintain high stability in both plasma and intracellular environments, remaining intact despite changes in pH, enzymatic action, or reducing agents. This means drug release depends entirely on the gradual degradation of the antibody in lysosomes, resulting in minimal free drug in circulation and lower systemic toxicity risk. Such stability is particularly suitable for highly toxic payloads that require strict release control.
| Linker Type | Description |
| Acid Cleavable Linkers | Design and synthesis of acid-sensitive linkers with high plasma stability and rapid cleavage in acidic tumor or lysosomal environments. |
| Disulfide Linkers | Development and optimization of disulfide linkers exploiting intracellular reducing conditions for selective and efficient payload release. |
| Cathepsin B Cleavable Linkers | Engineering of Cathepsin B–responsive linkers with precise peptide sequences for targeted drug activation in tumor cells. |
| Phosphatase Cleavable Linkers | Custom synthesis of phosphatase-sensitive linkers enabling selective drug release in enzyme-rich pathological tissues. |
| Sulfatase Cleavable Linkers | Design of sulfatase-cleavable linkers leveraging tissue-specific enzyme expression for localized payload delivery. |
| β-Galactosidase Cleavable Linkers | Development of β-galactosidase–responsive linkers with high enzymatic specificity for controlled drug release. |
| β-Glucuronidases Cleavable Linkers | Design of β-glucuronidase-cleavable linkers for site-specific drug liberation in tumor and inflammatory microenvironments. |
The release mechanism of the linker is one of the key factors in ADC design, determining whether the drug can act efficiently and precisely within target cells. Depending on the triggering signal, linker release modes can be categorized into enzyme-cleavable, pH-sensitive, glutathione-sensitive, and passive release types.
The design principle of enzyme-cleavable linkers is to take advantage of the high expression of specific enzymes inside tumor cells, so that the linker is recognized and cleaved by certain proteases after entering the target cell. A common example is the Cathepsin B-sensitive linker (such as the Val-Cit peptide), which is cleaved in the lysosomal environment by Cathepsin B, rapidly releasing the drug. In addition, some tumor tissues overexpress matrix metalloproteinases (MMPs), which can also serve as triggering mechanisms; by designing MMP-cleavable peptide sequences, precise drug release at tumor sites can be achieved. The advantage of this approach lies in its fast release speed and strong selectivity, but it is essential to ensure low enzyme levels in normal tissues to avoid off-target release.
pH-sensitive linkers primarily exploit the acidic characteristics of the tumor microenvironment and intracellular vesicles to trigger drug release. A typical example is the hydrazone linker, which rapidly breaks under acidic conditions (pH 4.5–6.0) while remaining stable in plasma at pH 7.4. Such linkers perform well in ADCs targeting endosomes or lysosomes, enabling rapid payload release after entering the cell. However, if the chemical stability of the linker is insufficient, partial hydrolysis may occur in plasma, leading to off-target toxicity. Therefore, researchers often modify the hydrazone structure to improve plasma stability while maintaining acid sensitivity.
Glutathione-sensitive linkers rely on the difference in redox environments between the inside and outside of cells to achieve drug release. The glutathione (GSH) concentration in the cytoplasm of tumor cells is typically 100–1000 times higher than in plasma, providing a natural triggering condition for disulfide linkers. Once an ADC enters a tumor cell, the disulfide bond is reduced to thiol groups under the high GSH concentration, breaking the linker and releasing the drug. This mechanism's advantage is that it effectively exploits the reductive difference between tumor and normal tissues; however, in certain diseases or physiological states, normal cell GSH levels may increase, raising off-target risks. To improve selectivity, some research teams introduce steric hindrance or modify the disulfide bond structure.
Non-cleavable linkers do not rely on external signals to break; instead, drug release occurs via proteolytic degradation of the antibody inside the cell. When an ADC enters the lysosome, the antibody scaffold is broken down into amino acid fragments, and the drug is released in complex with residual linker moieties. These residual linker–drug complexes generally retain cytotoxic activity and can effectively kill target cells. The advantage of non-cleavable linkers is their extremely high stability in plasma, with virtually no non-specific release, resulting in lower off-target toxicity. However, their release process is relatively slow, making them unsuitable for treatments requiring an immediate high-concentration drug attack.
In ADC design, linker selection is one of the critical factors determining drug stability and therapeutic performance. Different types of linkers have their own strengths and weaknesses in terms of release speed, plasma stability, and toxicity control. Researchers must balance these aspects based on the specific drug type, target characteristics, and clinical requirements.
Advantages: Cleavable linkers can rapidly break under specific biochemical conditions inside target cells, quickly releasing the drug. This rapid release helps achieve high cytotoxic concentrations in a short time, enhancing the inhibitory effect on target cells. For highly potent cytotoxic drugs that require rapid action (such as microtubule inhibitors and DNA-damaging agents), cleavable designs can maximize therapeutic potential. In addition, these linkers can leverage differences between tumor and normal cells in enzyme levels, pH, or redox states to achieve high selectivity.
Disadvantages: The high sensitivity of cleavable linkers also brings certain risks—if the design is not precise enough, non-target environments in circulation may trigger premature drug release. Such early release can raise the concentration of free drug in the bloodstream, causing systemic side effects. Furthermore, in some pathological conditions, normal tissues may exhibit elevated enzyme activity or redox levels, increasing off-target risks. Therefore, in clinical applications, careful evaluation of stability and triggering specificity is required.
Advantages: Non-cleavable linkers show extremely high chemical stability in plasma and do not easily break even inside cells. This design significantly reduces the risk of free drug leakage during circulation, lowering off-target toxicity. Since drug release mainly depends on proteolytic degradation of the antibody in lysosomes, the process is more controlled, helping maintain a longer duration of drug activity. For cytotoxic drugs that are structurally sensitive or unstable, such linkers provide strong protection.
Disadvantages: Because drug release depends on antibody degradation, non-cleavable linkers generally exhibit slower onset of action, which may limit efficacy in situations requiring rapid, high-intensity drug effects. Moreover, the released drug is often in the form of a linker–drug complex, which, while still cytotoxic, may have lower activity than the free drug. As such, design considerations must balance stability with release efficiency.
In ADC development, the properties of the payload and the biological characteristics of the target directly influence linker selection. For small-molecule chemotherapeutics that require rapid action—especially those with short half-lives or time-sensitive cytotoxic effects—cleavable linkers are more suitable, as they enable swift drug release inside target cells for strong cytotoxicity. For payloads requiring high stability, such as those that degrade or deactivate easily in the bloodstream, non-cleavable linkers can better ensure drug integrity and safety during delivery. In practice, development teams typically integrate pharmacokinetic, toxicological, and clinical indication data to make a comprehensive assessment of the appropriate linker type.
The type of linker is not merely a matter of chemical synthesis design; it also plays a decisive role in determining the pharmacokinetics, safety, and ultimate efficacy of ADCs in clinical settings. Different linkers influence the drug's biodistribution, release rate, and off-target risk, making them strategically important in the process of clinical translation.
The structure of the linker directly affects the stability and circulation time of the ADC in the body. Cleavable linkers can rapidly release the drug within target cells, resulting in a higher drug concentration at the tumor site—an advantage for quickly killing tumor cells. However, they require high stability in plasma to avoid premature release. Non-cleavable linkers, on the other hand, tend to extend the ADC's half-life in circulation and release the drug gradually after lysosomal degradation. This produces a smoother drug concentration curve, supporting sustained tumor suppression. Therefore, the pharmacokinetic differences between linkers must be matched to specific clinical objectives.
Linker design is closely tied to the safety profile of ADCs. If cleavable linkers are prematurely triggered in non-target tissues, they may cause severe systemic toxicities such as myelosuppression or liver damage. In contrast, non-cleavable linkers—due to their high stability in blood and normal tissues—can significantly reduce free drug concentrations, thereby lowering off-target toxicity. However, this does not mean they are entirely risk-free, as residual linker–drug complexes released during antibody degradation may still affect non-target cells in certain cases. Consequently, safety evaluations in preclinical studies must be tailored to the characteristics of different linker types.
Approved ADCs clearly demonstrate the successful application of different linker types. For instance, Adcetris® (Brentuximab Vedotin) employs a cleavable Val-Cit peptide linker, which utilizes lysosomal Cathepsin B cleavage to efficiently release the cytotoxic MMAE within target cells, thereby significantly enhancing efficacy. Kadcyla® (T-DM1), in contrast, uses a non-cleavable thioether linker (DM1-SMCC) to release the payload DM1 through antibody degradation, effectively reducing off-target toxicity. These successful cases show that both cleavable and non-cleavable linkers can achieve outstanding clinical outcomes when appropriately matched with drug properties and disease characteristics.
In recent years, linker technology has been advancing toward higher precision and safety. In the cleavable linker domain, research has shifted toward multi-trigger mechanisms (e.g., responding to both enzymatic and pH changes) to enhance tumor specificity. In the non-cleavable domain, more attention is being given to site-specific conjugation and payload residue optimization to improve the activity of released drugs. Additionally, AI-assisted design and computational chemistry are being introduced into linker development to predict stability and trigger efficiency, accelerating preclinical screening. These trends suggest that future linkers will be more intelligent, controllable, and adaptable to various antibodies and drugs.
BOC Sciences has long been deeply engaged in ADC technology development, with extensive research expertise and industrial capabilities in the design, optimization, and production of ADC linkers. We fully understand that linker stability, selectivity, and release mechanisms directly determine ADC efficacy and safety. Therefore, during development, we integrate chemical synthesis, pharmacological evaluation, and process scale-up to ensure every step meets the highest standards.
From cytotoxins to linkers, explore our cutting-edge products for your ADC project.
| Catalog | Name | CAS | Price | |
| BADC-01934 | Docosanedioic acid | 505-56-6 | Bulk Inquiry | |
| BADC-01135 | Fmoc-N-amido-PEG4-propionic acid | 557756-85-1 | Bulk Inquiry | |
| BADC-00415 | BCN-PEG4-NHS ester | 1702356-19-1 | Bulk Inquiry | |
| BADC-01530 | N-Succinimidyl 3-maleimidopropionate | 55750-62-4 | Bulk Inquiry | |
| BADC-00933 | DBCO-NHS ester | 1353016-71-3 | Bulk Inquiry | |
| BADC-00410 | 6-Azidohexanoic acid NHS ester | 866363-70-4 | Bulk Inquiry | |
| BADC-01147 | DSS Crosslinker | 68528-80-3 | Bulk Inquiry | |
| BADC-01192 | Sulfo-SMCC sodium | 92921-24-9 | Bulk Inquiry | |
| BADC-00382 | Bis-PEG1-NHS ester | 65869-64-9 | Bulk Inquiry | |
| BADC-01402 | (p-SCN-Bn)-NOTA | 147597-66-8 | Bulk Inquiry |
Explore our advanced tools and expertise for next-generation ADC research and development.
Potent cytotoxins designed for targeted antibody-drug conjugate therapy.
Stable, selective linkers enabling precise drug release in ADCs.
Integrated payload-linker solutions for efficient and targeted ADC delivery.
Targeted therapeutics combining antibodies with cytotoxic drugs for precision treatment.











 Loading......
Loading......