ADC linker design is one of the core aspects in the development of antibody–drug conjugates (ADCs). Linkers not only determine the stability of the drug in plasma but also directly influence the efficiency of intracellular payload release and therapeutic safety. An ideal linker should remain highly stable in the circulatory system to prevent premature toxin release that could cause systemic toxicity, while being capable of rapid cleavage under specific conditions (such as acidic environments, enzymatic action, or reducing conditions) once inside the target cell, thereby achieving precise drug delivery. As ADC applications expand in the treatment of cancer and immune diseases, linker design faces increasingly high chemical and biological requirements. This guide will systematically analyze the chemical principles, structural characteristics, development challenges, and optimization strategies of ADC linker design, offering researchers practical solutions and hands-on experience.
The core advantage of ADCs lies in their ability to deliver highly potent cytotoxic drugs precisely to tumor cells, with the linker playing a key role in enabling this targeted delivery. ADC linkers determine not only the stability of the drug during blood circulation but also the efficiency of payload release inside target cells and the binding properties of the antibody. As ADC development advances into clinical application, researchers have placed higher demands on linker chemical design, structural optimization, and trigger mechanisms.
Antibody–drug conjugates are a class of innovative therapeutics that combine targeted delivery with powerful cytotoxicity. They are composed of three main components: a monoclonal antibody (mAb), a cytotoxic drug (payload), and a linker. The linker, serving as a molecular bridge between the antibody and the drug, connects the two structurally and functionally determines the precision and efficiency of drug delivery.
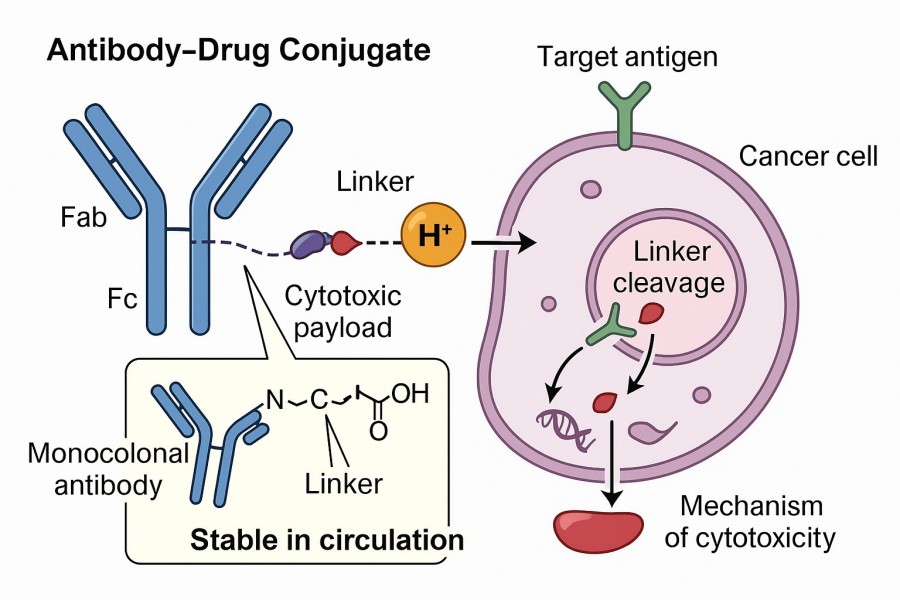 Fig. 1. Linker in antibody drug conjugate (BOC Sciences Authorized).
Fig. 1. Linker in antibody drug conjugate (BOC Sciences Authorized).
In ADC design, the linker is a critical structural unit that influences pharmacokinetics and tissue distribution, as well as the rate and location of drug release within target cells. Typical linker designs take into account reaction chemistry, molecular size, hydrophilic/hydrophobic balance, and chemical compatibility with both antibody and drug. Linkers must meet the following key functional requirements:
Linker performance directly dictates the clinical profile of an ADC. For example, studies have shown that ADCs with disulfide linkers can achieve rapid release in the reductive cytosol of tumor cells while remaining stable in plasma—significantly improving the therapeutic index.
ADC linker chemistry integrates principles from organic chemistry, biochemistry, and pharmacokinetics. Its primary objective is to maintain stability during circulation while enabling precise payload release in the tumor microenvironment. Successful designs account for bond stability, spatial conformation, hydrophilic/hydrophobic balance, molecular length, and cleavage mechanisms. The development process requires balancing plasma stability and intracellular release rate, ensuring compatibility with both payload and antibody, and validating performance through in vitro and in vivo testing.
Chemical stability
Controlled cleavability
Chemical compatibility
Pharmacokinetic tunability
Hydrophilic/hydrophobic balance
Optimizing molecular length
Choosing cleavage mechanisms
ADC linkers are classified into cleavable and non-cleavable types based on their cleavage mechanism, each with distinct differences in plasma stability, release method, payload compatibility, and antibody compatibility. Understanding these characteristics helps guide rational linker selection to maximize efficacy, minimize off-target toxicity, and meet clinical development needs.
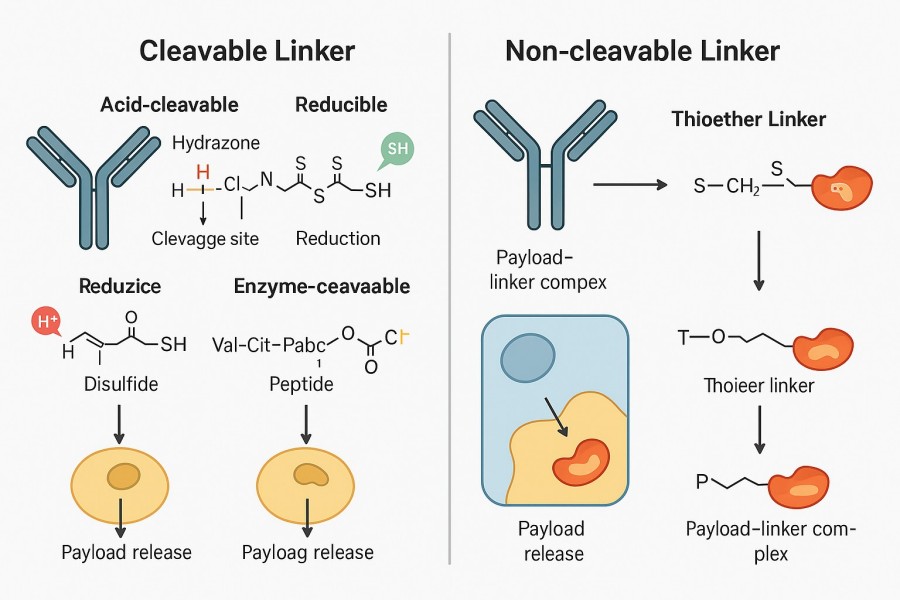 Fig. 2. Cleavable vs non-cleavable linkers (BOC Sciences Authorized).
Fig. 2. Cleavable vs non-cleavable linkers (BOC Sciences Authorized).
Cleavable linkers rely on specific intracellular conditions (pH, enzymes, reducing environment) to trigger cleavage and payload release. They are often used when rapid intracellular release of the free drug is desired.
Utilize the acidic environment of tumor cell lysosomes (pH 4.5–5.5) for selective cleavage. Common chemistries include hydrazone, acetal, and ketal bonds, which are stable at neutral plasma pH but hydrolyze rapidly under acidic conditions. They are straightforward to synthesize and were widely used in early ADCs. However, they may undergo slow hydrolysis in circulation, risking premature payload release and off-target toxicity. Example: Gemtuzumab ozogamicin (Mylotarg) uses a hydrazone linker to connect the antibody to calicheamicin, releasing the drug via acid hydrolysis inside target cells.
Depend on tumor-overexpressed proteases (e.g., Cathepsin B, Cathepsin L) to cleave peptide bonds and release the payload. Common sequences include Val-Cit and Ala-Val, often paired with self-immolative spacers such as PABC to ensure complete release. These linkers are highly stable in plasma but require adequate enzyme expression for efficient release. Example: Brentuximab vedotin (Adcetris) uses a Val-Cit-PABC linker cleaved by Cathepsin B in lysosomes to release MMAE.
Exploit the high glutathione (GSH) concentration and other reducing agents in the tumor cell cytosol for cleavage. Stable in normal plasma, but can be prematurely cleaved in mildly reducing environments. Steric hindrance (e.g., methyl or cycloalkyl groups) can be added to enhance plasma stability. These linkers enable rapid release without relying on enzyme systems, but their stability must be carefully controlled.
Non-cleavable linkers are designed to remain intact in both plasma and intracellular environments, releasing the payload only after the antibody is degraded in lysosomes. They often employ highly stable covalent bonds such as thioethers to ensure virtually no off-target release. The release product is typically a payload–linker residue conjugate rather than the free drug, limiting bystander effects but reducing collateral damage to healthy cells. This greatly improves safety and is particularly suitable for extremely potent payloads with well-defined intracellular targets. Example: Trastuzumab emtansine (Kadcyla) uses an SMCC linker, which is stable in plasma and releases a DM1–linker complex following antibody degradation inside target cells.
| Linker Type | Trigger Mechanism | Representative Drug | Advantages | Disadvantages | Suitable Payloads |
| Acid-labile | Hydrolysis in acidic lysosomal pH | Mylotarg | Simple synthesis, rapid release | Possible slow hydrolysis in plasma leading to off-target release | DNA-damaging agents, antimetabolites |
| Enzyme-cleavable | Protease-mediated cleavage | Adcetris | High selectivity, high stability | Dependent on protease expression levels | Microtubule inhibitors, toxins |
| Disulfide | Cleavage in reductive intracellular environment | Various experimental ADCs | Rapid release, broad applicability | Possible premature cleavage in plasma | Various cytotoxic agents |
| Non-cleavable | Release after antibody degradation | Kadcyla | Highest plasma stability, lowest off-target toxicity | No free drug release, limited bystander effect | Highly potent toxins, intracellular targets |
| Linker Type | Description |
| Acid Cleavable Linkers | Designed and synthesized with optimized acid sensitivity to ensure precise payload release in targeted acidic environments. |
| Disulfide Linkers | Customized with tailored stability and reduction-triggered cleavage properties for efficient intracellular drug release. |
| Cathepsin B Cleavable Linkers | Engineered with selective enzyme recognition sites for controlled release in cathepsin B–rich environments. |
| Phosphatase Cleavable Linkers | Developed with high substrate specificity to achieve regulated payload activation via phosphatase activity. |
| Sulfatase Cleavable Linkers | Constructed for targeted cleavage by sulfatases to enhance tumor-specific drug delivery. |
| β-Galactosidase Cleavable Linkers | Designed with optimized β-galactosidase–responsive moieties for selective activation in relevant biological contexts. |
| β-Glucuronidases Cleavable Linkers | Developed with enhanced enzymatic sensitivity to β-glucuronidases for effective tumor-localized payload release. |
Despite significant advances in modern ADC design, the development of linkers still faces numerous technical challenges, including balancing plasma stability with intracellular release, controlling off-target toxicity, and ensuring compatibility with both the payload and the antibody. Each of these challenges can impact the efficacy, safety, and manufacturability of an ADC. Through molecular modifications, structural optimization, and comprehensive analytical approaches, these difficulties can gradually be overcome.
One of the greatest challenges in ADC linker design is achieving both high stability in plasma and efficient release inside target cells. Overly stable linkers may result in insufficient release within tumor cells, reducing drug potency; conversely, insufficiently stable linkers may prematurely break in plasma, leading to off-target payload release and systemic toxicity.
To address this contradiction, researchers often employ refined chemical modifications to optimize trigger mechanisms. For example, acid-sensitive linkers can be tuned for pH sensitivity by introducing electron-donating or electron-withdrawing substituents, while enzyme-cleavable linkers can be designed with optimal peptide sequences (e.g., Val-Cit-PABC) to ensure cleavage only by specific proteases. Additionally, dual validation of in vitro stability and intracellular release efficiency—such as using human plasma stability assays and cell lysate release assays—is critical for ensuring successful design.
The clinical safety of ADCs largely depends on the linker's ability to release the payload selectively. If the linker releases the payload in normal tissues or the bloodstream, severe adverse effects can occur, including myelosuppression, liver injury, or peripheral neuropathy.
To reduce off-target toxicity, researchers employ highly specific trigger mechanisms (e.g., Cathepsin B–cleavable linkers) and use steric hindrance modifications (such as introducing methyl groups around disulfide bonds) to prevent nonspecific cleavage. Drug design may also incorporate bystander effect regulation: for highly heterogeneous tumors requiring strong cytotoxicity, cleavable linkers can be chosen to enable surrounding cancer cell killing; for therapies requiring precise targeting, non-cleavable linkers are preferred to avoid damage to non-target cells. Clinically, Kadcyla significantly reduced cardiac and hepatic toxicity by using a non-cleavable linker.
The chemical properties of a linker must be structurally and functionally compatible with the chosen payload and antibody; otherwise, issues such as low conjugation efficiency, poor drug stability, or reduced activity may occur. For instance, if a highly hydrophobic payload is directly conjugated to an antibody, ADC aggregation and reduced solubility may result, affecting in vivo distribution and efficacy.
Common strategies to address this problem include introducing hydrophilic modifying groups (such as PEG chains), adjusting linker length to avoid steric hindrance affecting antibody binding, and selecting conjugation chemistries with appropriate reactivity (e.g., maleimide–thiol coupling or click chemistry). It is also necessary to consider the chemical stability of the payload after release—if the released product is easily metabolized or inactivated, the linker design may need protective groups or drug structure modifications. For example, Brentuximab vedotin's Val-Cit linker is highly compatible with MMAE and avoids reduced antibody affinity.
To enhance ADC efficacy and safety, linker optimization strategies encompass chemical modifications, integration with antibody engineering and payload selection, and precise analytical evaluation methods. By adjusting linker hydrophilicity, length, cleavage mechanism, and spatial conformation, plasma stability and intracellular release efficiency can be optimized. Integrating linker design with antibody optimization and payload matching maximizes the therapeutic index of an ADC. Furthermore, accurate in vitro and in vivo analytical methods—such as plasma stability measurements, intracellular release kinetics assessments, and structural characterization—provide a scientific basis for optimization. Combining these strategies facilitates the creation of high-performance, clinically applicable ADC molecules.
Chemical modification is a core approach for optimizing linker performance. By altering bond types, introducing steric hindrance, or adjusting molecular hydrophilicity, plasma stability and intracellular release characteristics can be significantly improved. For example, in acid-sensitive linkers, the acid sensitivity of hydrazone bonds can be tuned by adding electron-donating or electron-withdrawing substituents to prevent slow hydrolysis in plasma; in disulfide linkers, introducing methyl or cycloalkyl steric groups effectively reduces the risk of nonspecific cleavage in plasma. In addition, adding hydrophilic modifications (such as short PEG chains) can adjust the solubility of hydrophobic payloads, prevent ADC aggregation, and improve drug distribution and circulation half-life.
Linker optimization must be closely integrated with antibody engineering and payload selection. The structure, glycosylation status, and surface charge of different antibodies can influence conjugation efficiency and overall ADC stability. Selecting appropriate conjugation sites (such as cysteine thiols or lysine amines) and matching them with the chemical properties of the payload can maximize conjugation efficiency while avoiding damage to antibody conformation or activity. For highly hydrophobic toxins such as MMAE, adjusting hydrophobicity through short PEG chains or polar linkers can reduce aggregation; for structurally sensitive payloads, mild conjugation conditions or click chemistry are preferred for high-fidelity conjugation. This integrated approach allows researchers to optimize linker performance while ensuring overall ADC stability and efficacy.
Implementing optimization strategies relies on precise analytical methods to verify linker performance. Common techniques include plasma stability testing, cell lysate release kinetics evaluation, mass spectrometry (LC-MS/MS), and high-performance liquid chromatography (HPLC) to monitor linker–drug conjugation states. These methods help quantify nonspecific cleavage rates in plasma, payload release efficiency inside cells, and conjugate homogeneity. Furthermore, in vitro toxicity tests and in vivo pharmacokinetic studies in animals can assess the impact of optimization strategies on the ADC's therapeutic index. Using these analytical tools in combination not only validates design feasibility but also helps identify potential issues early in development, reducing the risk of clinical failure.
ADC linker optimization is an iterative process involving cycles of design, synthesis, analysis, and validation. Each round of iteration is based on the results of the previous experiments, adjusting linker structures, conjugation strategies, or trigger mechanisms accordingly. For example, if initial plasma stability testing shows that an acid-sensitive linker hydrolyzes too quickly, researchers may modify electron substituents or peptide sequence length before conducting a second round of testing. This iterative approach, combined with high-throughput analytical techniques (such as microplate-based lysosomal-mimicking release assays) and computational molecular modeling, enables rapid identification of optimal linker designs, ultimately achieving efficient, stable, and safe ADC molecules.
With years of experience in ADC development and chemical synthesis, BOC Sciences offers comprehensive, customized linker design and development services for global clients. Our technical team includes experts in organic chemistry, biochemistry, pharmacokinetics, and analytical chemistry, enabling us to tightly integrate molecular design, chemical synthesis, conjugation processes, and quality control to deliver linker solutions with optimal stability and release characteristics. We focus not only on delivering individual products but also on providing systematic services that help clients address complex challenges in ADC development and accelerate projects from early research to clinical trials.
From cytotoxins to linkers, explore our cutting-edge products for your ADC project.
| Catalog | Name | CAS | Price |
| BADC-01147 | DSS Crosslinker | 68528-80-3 | Bulk Inquiry |
| BADC-01192 | Sulfo-SMCC sodium | 92921-24-9 | Bulk Inquiry |
| BADC-00409 | Azido-PEG2-NHS ester | 1312309-64-0 | Bulk Inquiry |
| BADC-01934 | Docosanedioic acid | 505-56-6 | Bulk Inquiry |
| BADC-00382 | Bis-PEG1-NHS ester | 65869-64-9 | Bulk Inquiry |
| BADC-00453 | Mal-PEG2-NHS ester | 1433997-01-3 | Bulk Inquiry |
| BADC-01919 | Boc-D-trans-Hyp-OH | 147266-92-0 | Bulk Inquiry |
| BADC-01112 | Fmoc-N-amido-PEG1-acetic acid | 260367-12-2 | Bulk Inquiry |
| BADC-00459 | SPDP-PEG8-NHS | 1252257-56-9 | Bulk Inquiry |
| BADC-00364 | Fmoc-Val-Cit-PAB | 159858-22-7 | Bulk Inquiry |
Explore our advanced tools and expertise for next-generation ADC research and development.
Potent cytotoxins designed for targeted antibody-drug conjugate therapy.
Stable, selective linkers enabling precise drug release in ADCs.
Integrated payload-linker solutions for efficient and targeted ADC delivery.
Targeted therapeutics combining antibodies with cytotoxic drugs for precision treatment.











 Loading......
Loading......