ADC manufacturing is an emerging and highly complex biopharmaceutical technology that has attracted significant attention from innovative biopharmaceutical companies and CDMO procurement professionals. Antibody–drug conjugates (ADCs) are a class of precision therapeutics in which highly potent small-molecule cytotoxic drugs are chemically linked to tumor-targeting antibodies. The design concept originates from the "magic bullet" theory, combining the specificity of monoclonal antibodies with the powerful cytotoxicity of highly toxic drugs, enabling safe delivery of the drug to tumor cells and selective release of the toxic component. As an increasing number of ADCs enter clinical development, a thorough understanding of ADC manufacturing processes, key quality attributes, and regulatory requirements is critical for biopharmaceutical developers.
The manufacturing of ADCs is a highly interdisciplinary process that integrates biopharmaceutical production, small-molecule chemistry, and advanced analytical technologies. The ADC manufacturing process includes several key steps, such as monoclonal antibody expression and purification, synthesis of highly potent payloads, and chemical linker conjugation. Compared with traditional biologics manufacturing (such as unconjugated monoclonal antibodies), ADC production must additionally address the safe synthesis of highly potent active pharmaceutical ingredients (HPAPIs) and the special requirements of conjugation conditions, resulting in higher process complexity and more stringent quality control. Due to the involvement of highly potent small molecules and the complexity of conjugation technologies, many companies choose to outsource ADC projects to experienced CDMOs to reduce risk and accelerate product development.
ADCs are typically composed of three core components: a targeting antibody, a chemical linker, and a cytotoxic payload.
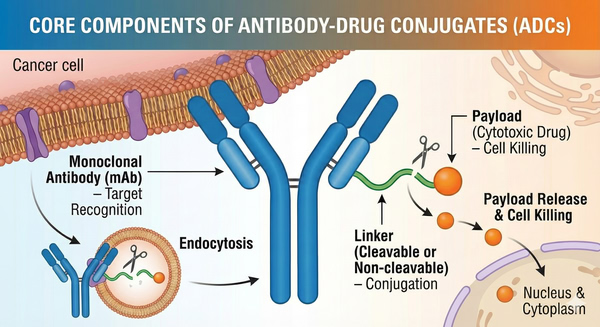 Fig. 1. The core components of an antibody drug conjugate (BOC Sciences Authorized).
Fig. 1. The core components of an antibody drug conjugate (BOC Sciences Authorized).
For example, linkers may be designed to hydrolyze under low-pH conditions or to be cleaved by specific intracellular enzymes, ensuring that the drug is released at the desired site. Ideal payload characteristics include extremely high potency, low immunogenicity, and the presence of reactive functional groups for linker attachment. Common payloads include microtubule inhibitors (such as MMAE and DM1) and DNA-damaging agents (such as acridine-based conjugates), whose cytotoxicity is typically 100–1000 times greater than that of conventional chemotherapeutic drugs.
In summary, antibody selection must balance stability, internalization rate, and immunogenicity; linker design must balance in vivo stability with efficient intracellular release; and payloads must possess sufficient potency while allowing safe and controlled conjugation.
ADC manufacturing differs significantly from traditional monoclonal antibody production and presents additional challenges. First, in addition to standard antibody expression and purification, ADCs require the separate synthesis of highly potent cytotoxic payloads and the design of suitable linkers for conjugation. This means that ADC projects demand coordinated development of both macromolecules (antibodies) and small molecules (payloads), as well as expertise in chemical conjugation and toxicological evaluation.
Moreover, due to the extreme toxicity of payloads, ADC processes must be carried out in closed systems or multi-level containment environments to ensure operator safety. From a quality control perspective, beyond routine assessments of protein purity and activity, unique parameters such as the drug-to-antibody ratio (DAR) must be carefully monitored. As a result of this high technical complexity, many developers choose to collaborate with specialized ADC CDMOs and outsource their projects to accelerate clinical development and commercialization.
The ADC manufacturing workflow typically includes the following major stages: monoclonal antibody production and purification, synthesis and handling of cytotoxic payloads, linker design and chemical conjugation, purification of conjugates and removal of free drug, and final formulation and fill-finish. Each stage is closely interconnected and collectively determines product quality and accessibility. The key process steps of each stage are described below:
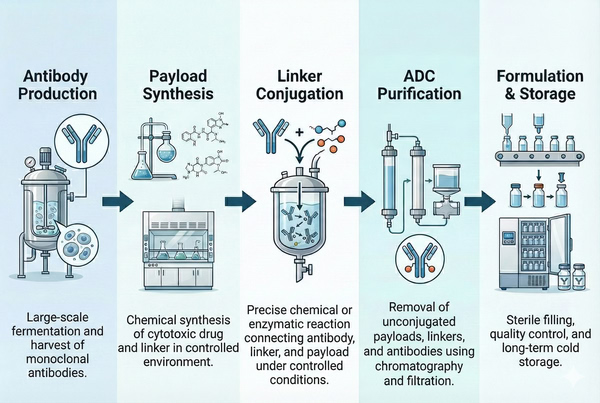 Fig. 2. Overview of the complete ADC manufacturing workflow, from antibody production to final formulation and storage (BOC Sciences Authorized).
Fig. 2. Overview of the complete ADC manufacturing workflow, from antibody production to final formulation and storage (BOC Sciences Authorized).
Upstream cultivation: High-quality antibody genes must first be obtained and stably expressed. Cell lines (typically CHO cells) are expanded from seed cultures in the laboratory and then scaled up to large-scale fermentation in bioreactors. During this process, culture media, feeding strategies, temperature, and pH conditions must be optimized to achieve high antibody yields and consistent quality.
Downstream purification: After fermentation, cell debris is removed through centrifugation and filtration, followed by sequential chromatographic purification. Common purification schemes include Protein A affinity chromatography to capture antibodies, followed by ion-exchange and hydrophobic interaction chromatography to remove host-cell impurities, and final sterile filtration to obtain purified antibodies. The high-purity antibody produced at this stage serves as the starting material for subsequent conjugation.
Antibody modification: In ADC processes, purified antibodies often require chemical or enzymatic modification, such as introducing reactive functional groups at glycan moieties or specific sites on the antibody to enable subsequent payload conjugation. Strict process monitoring is required throughout this stage to ensure antibody stability and preservation of biological activity.
Cytotoxic payloads (HPAPIs) are typically extremely potent small-molecule compounds with special requirements for synthesis and handling. Payloads may be produced through chemical synthesis or semi-synthetic routes and are often synthesized and purified in dedicated high-containment facilities. These synthetic routes must be carefully designed, optimized, and validated. The final payload must meet very high purity standards (typically >99%) and contain the functional groups required for conjugation, while transportation and storage must be strictly controlled to prevent exposure. Throughout the entire process, operator safety is of paramount importance, requiring multiple protective measures such as ventilation control, contained operations, and specialized training.
The role of the linker is to stably attach the payload to the antibody while allowing drug release in the target intracellular environment. Linkers are generally classified as cleavable or non-cleavable. Cleavable linkers utilize mechanisms such as acid-labile bonds, disulfide bonds, or peptide sequences that are cleaved under specific intracellular conditions to release the drug; non-cleavable linkers (such as cyclohexane maleimide structures) require complete lysosomal degradation of the antibody to release the active metabolite. Linkers are often further modified with hydrophilic groups (such as PEG) to improve ADC solubility and stability.
In terms of conjugation chemistry, traditional methods rely on covalent attachment to cysteine thiols or lysine amines on antibodies, which results in relatively random conjugation. In recent years, site-specific conjugation technologies have emerged, enabling payload attachment at predefined antibody sites to produce more homogeneous products. Common strategies include engineering antibodies to introduce additional cysteine residues (such as THIOMAB technology), enzymatic conjugation using native glycans or specific peptide sequences, and bioorthogonal conjugation using non-natural amino acids. For example, GlycoConnect™ technology first employs endoglycosidases to trim antibody glycans and uses glycosyltransferases to introduce azide functional groups; subsequently, a metal-free cyclooctyne–azide "click" reaction is used to conjugate a cyclooctyne-containing drug linker to the antibody. This two-step enzymatic–click approach enables the production of highly homogeneous ADC molecules.
After chemical conjugation, the reaction mixture contains unconjugated antibodies (DAR 0), conjugates with different DAR values, free drug, and linker-related impurities. To obtain high-purity ADC products, multiple downstream purification methods are employed. For example:
Through these multidimensional purification processes, free toxins, unconjugated antibodies, and other small-molecule impurities are removed, yielding ADC formulations that meet quality standards.
After purification, ADCs must undergo final formulation and sterile fill-finish to be prepared as drug products. During formulation, appropriate buffer systems, pH values, and excipients are selected based on the physicochemical properties of the ADC (such as hydrophobicity and isoelectric point) to enhance solubility and stability. Common approaches include adding low concentrations of organic solvents or surfactants, as well as anti-aggregation agents and antioxidants, to prevent aggregation or degradation of hydrophobic ADCs. Following formulation, the product is sterilized through 0.22 μm filtration and filled under aseptic conditions. Glass vials or prefilled syringes are commonly used, and fill-finish equipment and filters are subjected to integrity testing and sterility assurance to ensure compliance with GMP requirements. After filling, ADC formulations may be stored frozen or refrigerated, with storage conditions and shelf life determined based on stability and real-time stability studies.
Discover our comprehensive conjugation and manufacturing services, from initial process development to efficient scale-up. Engineered for high-purity yields, robust batch-to-batch consistency, and seamless technology transfer.
Successful ADC development relies heavily on multiple key manufacturing technologies that collectively determine product homogeneity, safety, and clinical developability. As ADC pipelines grow in complexity and quality requirements become more stringent, manufacturing processes increasingly incorporate more precise, controllable, and scalable technologies to meet strict demands for DAR control, process reproducibility, and large-scale production. Several commonly used key technology platforms are described below:
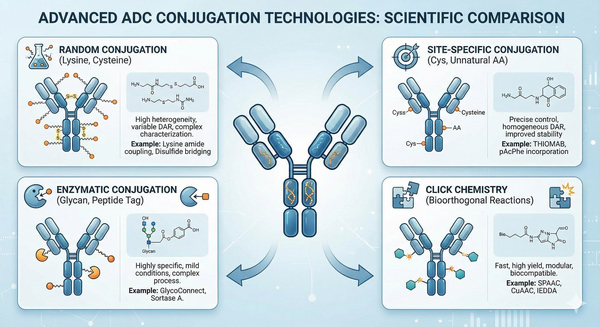 Fig. 3. Overview of key conjugation technologies applied in ADC manufacturing (BOC Sciences Authorized).
Fig. 3. Overview of key conjugation technologies applied in ADC manufacturing (BOC Sciences Authorized).
Traditional conjugation methods typically employ non-specific chemical modification of lysine or cysteine residues, resulting in heterogeneous ADC mixtures with multiple DAR values and conjugation sites. Site-specific conjugation technologies, by contrast, use engineered antibodies or specific enzymatic reactions to precisely control conjugation sites, producing more homogeneous ADCs. For example, introducing a defined number of cysteine residues (THIOMAB technology) enables the generation of ADCs with near-uniform DAR values.
Enzymatic conjugation approaches use antibody glycans or short peptide sequences as anchoring points to introduce special functional groups through enzymatic reactions, followed by conjugation via click chemistry. For instance, GlycoConnect™ technology enzymatically modifies antibody glycans to introduce azide-functionalized sugars, which are then conjugated to cyclooctyne-bearing drug linkers through strain-promoted azide–alkyne cycloaddition (SPAAC). This chemo-enzymatic approach offers high controllability and excellent product uniformity.
Site-specific conjugation strategies enable precise control of the average DAR. Studies have shown that ADCs typically achieve optimal antitumor efficacy at DAR values of 2–4, while excessively high DARs increase aggregation and clearance. For example, engineered cysteine conjugation allows for fixed DAR values (such as 2 or 4), resulting in more consistent DAR distributions across batches, reduced toxicity, and improved therapeutic efficacy.
Because ADC manufacturing involves highly toxic materials, closed production environments are essential. Single-use systems (such as disposable bioreactor bags and tubing) reduce cross-contamination risks and eliminate complex cleaning procedures, improving safety and flexibility. Modern ADC manufacturing facilities typically employ multiple physical containment measures, including isolators, air showers, and negative-pressure control, to protect personnel and ensure product quality.
Although ADC manufacturing technologies continue to evolve, the production process still faces multiple technical and engineering challenges across process development, quality control, and scale-up. The safe handling of highly potent payloads, product heterogeneity arising from conjugation reactions, and insufficient process reproducibility can all directly affect ADC manufacturability and clinical progression. In response to these critical challenges, the industry is addressing them through process optimization, advanced conjugation technologies, and systematic quality control strategies.
The HPAPIs used in ADCs are extremely toxic, imposing stringent requirements on production environments and personnel safety. To mitigate risks, manufacturing facilities must be equipped with high-level containment measures, comprehensive ventilation systems, and specialized personal protective equipment. Wherever possible, closed or automated systems are employed, and the use of single-use technologies further reduces operator exposure. In addition, dedicated safety training for manufacturing personnel and the implementation of strict operating procedures are of paramount importance.
The inherent heterogeneity introduced during conjugation leads to variability in DAR species and conjugation site distribution, which can result in inconsistent product performance. To address this issue, site-specific conjugation technologies can be adopted to improve ADC homogeneity. In parallel, downstream purification techniques such as HIC are used to separate and analyze different DAR species, enabling strict control of the final product's DAR distribution.
Conjugation efficiency is highly sensitive to conditions such as pH, temperature, and reactant concentration, making yield variability a common challenge. To overcome this, reaction condition screening and process optimization are essential to improve conjugation efficiency, along with the development of in-line monitoring and end-point detection methods. For example, companies such as BOC Sciences design systematic reaction screening and establish DAR control strategies during conjugation development to ensure reproducibility from laboratory scale to larger-scale manufacturing.
Scaling ADC processes from the laboratory to clinical and commercial production requires maintaining consistent product quality attributes while addressing challenges related to the supply of highly potent materials and equipment safety. During scale-up, purification performance and product stability must be repeatedly verified, and process parameters may need adjustment at different scale levels. Therefore, a robust HPAPI supply chain and experienced process engineers are critical to successful ADC scale-up.
To ensure that products meet clinical and regulatory requirements for safety, efficacy, and uniformity, a wide range of advanced analytical methods is applied throughout the manufacturing process. These methods focus on monitoring critical quality attributes such as DAR, free drug content, structural integrity, and stability. Systematic analytical strategies not only safeguard product quality but also provide a scientific basis for process optimization and scale-up.
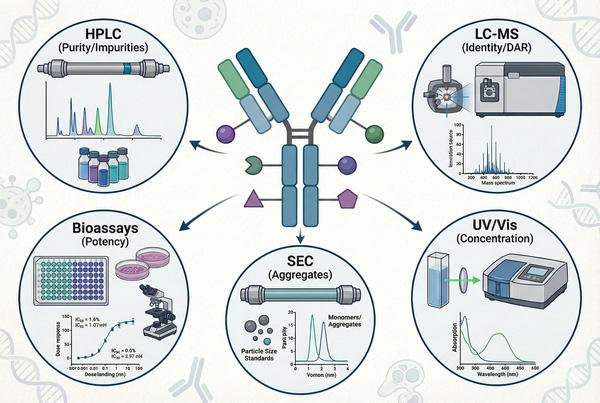 Fig. 4. Key analytical methods used for characterization and quality control of ADCs (BOC Sciences Authorized).
Fig. 4. Key analytical methods used for characterization and quality control of ADCs (BOC Sciences Authorized).
The average DAR is a key quality attribute of ADCs and is commonly analyzed using hydrophobic interaction chromatography (HIC) to separate and profile different DAR species. In addition, UV–visible spectroscopy (based on the distinct absorbance characteristics of the antibody and payload) or liquid chromatography–mass spectrometry (LC-MS) can be used for accurate quantification to determine the average DAR and the relative proportions of different DAR species.
Analysis of unconjugated free drug, free linker, and other low-molecular-weight impurities in the final formulation is critical. High-performance liquid chromatography (HPLC) and related techniques are typically used to detect free payloads and their derivatives. Strict impurity specifications and high-sensitivity analytical methods ensure that free toxins remain below established safety limits.
Mass spectrometry, peptide mapping, and glycan analysis are employed to verify the structural integrity of ADCs, along with accelerated and long-term stability studies. These evaluations assess ADC stability under different pH, temperature, and light conditions, ensuring that the final product maintains its activity and purity throughout its shelf life.
In addition to chemical analyses, ADCs are routinely evaluated using in vitro cytotoxicity assays and pharmacodynamic studies to confirm biological activity. In vitro release assays simulate drug release behavior under target-relevant conditions, assessing linker performance and payload release efficiency. Although this article focuses on manufacturing technologies, it should be noted that these biological assays are also an integral part of the overall ADC quality evaluation framework.
Because ADCs combine characteristics of both biologics and small-molecule drugs, their development and manufacturing are considered highly complex from a regulatory perspective. Major global regulatory authorities, including the FDA, EMA, and China's NMPA, have established clear technical expectations for ADC products in pharmaceutical development, process control, and quality evaluation. These regulatory requirements emphasize adherence to existing frameworks for biologics and chemical drugs, while also calling for more systematic and controlled strategies to address ADC-specific factors such as conjugation processes, highly potent payloads, and product heterogeneity. In general, ADC development requires additional focus on the following key considerations beyond standard regulatory requirements:
Because ADC manufacturing involves highly toxic payloads and intermediates, global regulatory agencies generally require GMP facilities to demonstrate dedicated capabilities for handling highly potent materials. Manufacturing sites used for ADC production typically include segregated containment areas designed to accommodate substances with different occupational exposure bands (OEBs), and to minimize personnel and environmental exposure through multiple layers of engineering controls. Common facility designs include negative-pressure control, airlock systems, and closed or isolated operation units to ensure that highly potent materials are safely and consistently managed throughout the production process, in line with international GMP and occupational safety expectations.
Comprehensive technical documentation and validation data are required throughout the entire manufacturing lifecycle. Each ADC batch must be supported by complete process validation reports, batch production records, and certificates of analysis (COAs) for regulatory submissions and audits. Process validation must cover critical steps such as conjugation and purification, demonstrating that the process is reproducible and well controlled. In addition to standard purity and activity testing, release criteria must include ADC-specific parameters such as DAR and free toxin levels.
Regulatory authorities typically require a comprehensive evaluation of ADC safety and efficacy, including biodistribution studies, stability monitoring, and validation of manufacturing processes and analytical methods. During the preclinical stage, emphasis is placed on process feasibility and consistency; for IND and BLA submissions, detailed CMC (Chemistry, Manufacturing, and Controls) documentation is required. Overall, ADC product approval follows established biologics guidelines while placing particular emphasis on the unique challenges associated with high toxicity and conjugation technologies.
From discovery to clinical submission, ADC programs undergo multiple rounds of process development and progressive scale-up. At each scale transition, conjugation homogeneity, DAR, and product stability must be maintained, while also addressing operational safety and the specialized handling requirements of highly potent payloads.
In early development stages, teams typically conduct small-scale (milligram to gram) process optimization to identify optimal conjugation conditions and formulations. Organizations such as BOC Sciences perform reaction condition screening, DAR design, and preliminary sample preparation at this stage. Parameters such as conjugation buffer, temperature, and drug loading are optimized to establish a robust manufacturing process and generate research-grade materials required for IND submission.
As ADCs advance into clinical trials, production must be scaled up to meet clinical dosing requirements (often at the tens-of-liters scale), accompanied by supplemental validation. Following successful clinical batches, commercial manufacturing requires further scale-up to hundreds of liters or more. Throughout this process, scalability of critical parameters—such as mixing, mass transfer, and in-process control—must be maintained, while ensuring compliance with equipment and safety requirements. For example, HPAPI intermediate production may initially require only hundreds of grams, whereas commercial manufacturing may demand kilogram-scale quantities, necessitating corresponding upgrades to supply chains and manufacturing facilities.
At each critical milestone, technology transfer is a key priority. Technology transfer involves transferring laboratory-developed processes, formulations, and analytical methods to GMP manufacturing teams, along with necessary engineering scale-up verification. This typically includes the preparation of technology transfer plans, review of raw materials and process documentation, and scale-up and reproducibility verification at the receiving site. Successful technology transfer is essential to ensuring that compliant ADC products can be consistently manufactured in new production environments.
As a professional ADC CDMO, BOC Sciences offers comprehensive service capabilities covering all stages of ADC development and manufacturing. Its ADC service portfolio includes integrated solutions ranging from molecular design, payload and linker synthesis, and conjugation process development to analytical testing and cGMP manufacturing. BOC Sciences operates cGMP facilities totaling more than 4,000 m² and is equipped with multiple standard isolators, enabling end-to-end support from DNA construction and cell expression to ADC cGMP production. In addition, the company has established robust analytical platforms and quality systems to accurately determine DAR and drug distribution, and to provide complete batch records and quality reports for each product, supporting regulatory submission and commercialization requirements.
From cytotoxins to linkers, explore our cutting-edge products for your ADC project.
| Catalog | Name | CAS | Price |
| BADC-00346 | Maytansine | 35846-53-8 | Bulk Inquiry |
| BADC-00033 | PNU-159682 | 202350-68-3 | Bulk Inquiry |
| BADC-01147 | DSS Crosslinker | 68528-80-3 | Bulk Inquiry |
| BADC-01192 | Sulfo-SMCC sodium | 92921-24-9 | Bulk Inquiry |
| BADC-00009 | DM1-SMCC | 1228105-51-8 | Bulk Inquiry |
| BADC-00008 | Val-Cit-PAB-MMAE | 644981-35-1 | Bulk Inquiry |
| BADC-00606 | Deruxtecan | 1599440-13-7 | Bulk Inquiry |
| BADC-00020 | DM1-SMe | 138148-68-2 | Bulk Inquiry |
| BADC-00972 | Fmoc-Gly-Gly-Phe-OH | 160036-44-2 | Bulk Inquiry |
| BADC-00790 | Mithramycin A | 18378-89-7 | Bulk Inquiry |
Explore our advanced tools and expertise for next-generation ADC research and development.
Potent cytotoxins designed for targeted antibody-drug conjugate therapy.
Stable, selective linkers enabling precise drug release in ADCs.
Integrated payload-linker solutions for efficient and targeted ADC delivery.
Targeted therapeutics combining antibodies with cytotoxic drugs for precision treatment.











 Loading......
Loading......